


Autism is the prototypical form of a group of pervasive developmental disorders that comprise autism, atypical autism, Asperger syndrome and ‘other’ pervasive developmental disorder. These conditions, as defined in ICD–10, are characterised by qualitative impairments in reciprocal social interaction, qualitative impairments in communication and stereotyped/repetitive patters of interest and activity. 1 An association between autism and epilepsy has long been recognised and is now well established. Prevalence estimates have varied, but between 11 and 39% of individuals with autism have been reported to develop epilepsy. Reference Kawasaki, Yokota, Shinomiya, Shimizu and Niwa2–Reference Tuchman, Moshe and Rapin7 The frequency of epilepsy is much higher than the prevalence of active epilepsy in the general population, which has been estimated in a UK epidemiological study to be 0.63% at age 23. Reference Kurtz, Tookey and Ross8 A similar increase in the prevalence of epilepsy has been reported in individuals with intellectual disability. Reference Goulden, Shinnar, Koller, Katz and Richardson9,Reference Matthews, Weston, Baxter, Felce and Kerr10
Despite the strong and well-established link, the basis for the association is poorly understood. The lack of progress in clarifying this stems from the presence of three major methodological limitations that have hampered progress in the identification of risk markers and factors. First, investigators have included various diagnostic subtypes in their reports of epilepsy in autism-spectrum disorder, including individuals with Rett syndrome and childhood disintegrative disorders (where the rate of epilepsy is over 80–90%), as well as including individuals with ‘other’ forms of pervasive developmental disorder. Reference Danielsson, Gillberg, Billstedt, Gillberg and Olsson11 Accordingly, the pattern of findings has been quite variable. Second, although individuals with autism sometimes do not develop epilepsy until late in adolescence, Reference Kawasaki, Yokota, Shinomiya, Shimizu and Niwa2,Reference Hara12 this age-related risk has not been taken into account in the design of many studies or the data analysis. Third, the studies conducted to date have not explored whether the familial liability for autism is correlated with the familial liability for epilepsy. This would be a test of the hypothesis that the genetic and environmental risk factors for autism also confer a risk for epilepsy.
In this study, we aimed to address some of the shortcomings of previous research by examining the correlates of epilepsy in a sample of individuals with autism who have been followed-up into adulthood. We have also collected family history data on the presence of autism and the broader phenotype of autism as well as epilepsy in first-, second- and third-degree relatives of a subset of these participants. These data have been analysed using survival-analysis techniques in order to map the correlated features of epilepsy in autism and to test the hypothesis that the liability to the familiality of autism confers an increased risk for epilepsy.
Method
Sample
All participants were originally seen at a specialist out-patient clinic for children with possible autism at the Maudsley Hospital between the years 1950 and 1985 inclusive. They were selected at that time to participate in a family study Reference Bolton, Macdonald, Pickles, Rios, Goode and Crowson13 and in a follow-up study. Reference Howlin, Goode, Hutton and Rutter14,Reference Hutton, Goode, Murphy, Le Couter and Rutter15 In order to be included in either study the participants had to have a diagnosis of autism or Asperger syndrome according to ICD–10 criteria. 1 This was established by an expert clinical evaluation. Diagnoses were confirmed by early versions of semi-standardised instruments (Autism Diagnostic Interview – Revised Reference Lord, Rutter and Le Couteur16 (ADI–R) and Autism Diagnostic Observational Schedule – Generic Reference Lord, Rutter, Goode, Heemsbergen, Jordan and Mawhood17 (ADOS–G)). Exclusion criteria included a history of infantile spasms, institutional rearing and severe/profound intellectual disability. Family study participants were stratified by gender and intellectual level. Ethical approval for the study was obtained from the local research ethics committee.
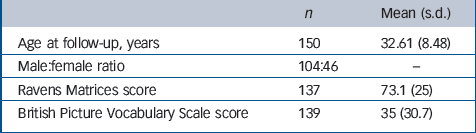
In total, 175 individuals of at least 21 years of age were eligible for the study (99 of them originated from the family study of Bolton et al Reference Bolton, Macdonald, Pickles, Rios, Goode and Crowson13 ). Our sample comprises participants previously described by Howlin, Rutter and associates. Reference Bolton, Macdonald, Pickles, Rios, Goode and Crowson13–Reference Hutton, Goode, Murphy, Le Couter and Rutter15 Follow-up data were available for 150 of these individuals; the remaining 25 were untraceable or did not respond to our follow-up letters (seeTable 1 for details of cohort). Families were approached in writing to request their participation and parents were sent a questionnaire that covered a range of issues Reference Hutton, Goode, Murphy, Le Couter and Rutter15 and specifically asked whether their son/daughter had ‘ever suffered from fits, seizures or epilepsy’ and if so what age the fits began, and whether the individual was taking medication for epilepsy. In addition, information about possible fits or seizures was available from answers to a question in the Autism Diagnostic Interview that was conducted when the individuals were first recruited into the study. Families/carers were then approached for further information regarding any individual with suspected fits or seizures and a detailed epilepsy interview (see below) was carried out. The age range of the participants at follow-up was 26–56 years (mean 32.61, s.d. = 8.48) (Table 1).
Table 1 Sample characteristics
| n | Mean (s.d.) | |
|---|---|---|
| Age at follow-up, years | 150 | 32.61 (8.48) |
| Male:female ratio | 104:46 | – |
| Ravens Matrices score | 137 | 73.1 (25) |
| British Picture Vocabulary Scale score | 139 | 35 (30.7) |
For the purpose of this study, we defined epilepsy as one or more non-febrile seizures that were not confined to the pre-school period (up to 5 years of age). The classification of seizure type followed the 1993 definitions of the International League Against Epilepsy. 18
Measures
Epilepsy
Information about the presence and characteristics of seizures was obtained using a semi-structured interview conducted by I.C-R. and P.F.B. The interview schedule was designed to help in the differential diagnosis of seizure disorders and systematically enquired about each type of seizure, its frequency and duration, as well as any possible precipitants (including prescribed medication), antecedents and post-ictal behaviours. Whenever possible, clinical records of the individuals were also examined. A clinical consensus on whether the reported epileptic seizures were in keeping with our definition of epilepsy was achieved among I.C-R., P.F.B. and M.R. Wherever possible we sought reports of electroencephalogram (EEG) investigations that had been undertaken in order to determine the nature of the type of epilepsy. However, because of the long follow-up interval, EEG data were only available for 18 individuals.
Developmental and cognitive assessments
Items from the ADI–R relating to language development were extracted for analyses. These included: ‘loss of language skills’–defined as a loss of language skills that lasted for at least 3 months after the child had achieved communicative use of at least five words (other than ‘mama’ or ‘dada’) on a daily basis for at least 3 months; and ‘overall level of language’–defined as functional use of spontaneous, echoed or stereotyped language on a daily basis that involved phrases of three words or more and that at least sometimes included a verb.
Cognitive assessments of these individuals have been conducted on a number of occasions over the years. For the purpose of this study we selected measures that were available for the largest number of participants. These included the Ravens Matrices Reference Raven, Court and Raven19 and the British Picture Vocabulary Scale (BPVS). Reference Dunn, Whetton and Pintillie20 Scores from these tests were used to construct ranked estimates of non-verbal (Ravens Matrices) and verbal ability (BPVS). An overall rating of verbal ability was obtained from the item coding overall level of language in the ADI–R.
Physical examination and obstetric complications
The obstetric histories of the individuals were previously collected, as part of the original family study, using the Obstetric Enquiry Schedule (OES) that was constructed using items from the Rochester Obstetric Scale Reference Zax, Sameroff and Babigian21 and the Gillberg Optimality Scale. Reference Gillberg and Gillberg22 Further information on the design of the OES can be found in Bolton et al. Reference Bolton, Murphy, Macdonald, Whitlock, Pickles and Rutter23 Data on the presence of minor congenital abnormalities were collected using the Waldrop and Halverson Scale. Reference Waldrop, Pedersen and Bell24
Use of psychotropic medications
In addition to information about possible fits/seizures, the screening questionnaire also asked if any new problems (in terms of major aggression, psychiatric illness or a marked escalation of long-standing problems) had emerged since the last contact with the family and for details about these problems. Reference Hutton, Goode, Murphy, Le Couter and Rutter15 For all individuals where information suggested a possible new-onset psychiatric disorder, further information was obtained by interviewing an informant who knew the person best, using a detailed interview schedule (Schedule for the Assessment of Psychiatric Problems in Autism, SAPPA). Reference Hutton, Goode, Murphy, Le Couter and Rutter15,Reference Bradley and Bolton25 This interview included questions about the individual's medication history and whether they had ever been prescribed major tranquilisers or antidepressants. The answers to the screening questionnaire and the information from the SAPPA and epilepsy interview were reviewed in order to determine who had been prescribed major tranquilisers (chlorpromazine, thioridazine, sulpiride, risperidone, etc.) or antidepressants. It was not usually possible to determine when these medications were prescribed, just whether they had been.
Family data
Data on family history were collected at an interview with a parent when the children were first enrolled in the study, using the Family History Interview (FHI). Reference Bolton, Macdonald, Pickles, Rios, Goode and Crowson13 The FHI is an investigator-based standardised instrument that documents the presence of developmental disorders of speech, reading and spelling; autistic symptoms (including abnormalities in social/communication development and repetitive behaviours and circumscribed interests); neurological (including seizure) and psychiatric disorders. The information from the interview is used to index the presence of the broader autism phenotype in first-, second- and third-degree relatives. Reference Bolton, Macdonald, Pickles, Rios, Goode and Crowson13
Statistical analysis
All analyses were carried out using STATA version 10 for Windows. Analysis of variance was used to compare means, and non-parametric tests (for example Mann–Whitney U-test) were used where appropriate. Survival analysis was undertaken using Cox's regression to estimate the proportional hazard and survival function. Family data were analysed using survival analyses and the cluster subcommand to take account of the fact that relatives from a family do not represent independent observations and to obtain robust estimates of variance. Logistic regression with the cluster subcommand was used to predict the determinants of the familial aggregation of the broader autism phenotype.
Results
The characteristics of the sample are summarised inTable 1. The male:female ratio was 2.3:1.
Prevalence and type of epilepsy
Of 150 individuals, 33 (22%) were found to have epilepsy as defined by our criteria. For 12 participants, queries were raised about possible epilepsy but they failed to meet our criteria (i.e. seizures were confined to the pre-school period or only occurred in the context of febrile illnesses or the features of the ‘fit’ were not clearly epileptiform). The lifetime prevalence of epilepsy (22%) was significantly higher than the lifetime prevalence of epilepsy reported in the 1958 British National Cohort Reference Kurtz, Tookey and Ross8 by age 23 years (0.7–1%; binomial test P<0.0001).
Detailed clinical seizure histories were available for 29 of the 33 participants. These histories were consistent with generalised tonic–clonic seizures in 88% of individuals. In four of these the history was consistent with partial seizures with secondary generalisation. The possibility of simple or complex partial epilepsy was raised in eight other individuals but was not confirmed in any of them. Three individuals (9.1%) had a history of febrile seizures that preceded the development of epilepsy.
We obtained EEG reports for 18 individuals who met our criteria for epilepsy. The EEG studies were undertaken as part of their epilepsy assessment. The EEG information for nine participants specifically reported on the nature of epileptic discharges and helped in the classification of seizure disorder type. In four individuals the EEG was reported to be abnormal, but did not contribute to seizure classification. In five individuals, the EEG was not reported to be abnormal.
In four participants the inter-ictal EEG revealed focal or multifocal spike wave discharges that might represent an epileptic focus, but the clinical descriptions of the seizures did not suggest the presence of partial seizures. Accordingly, we classified these individuals as just having generalised tonic–clonic seizures.
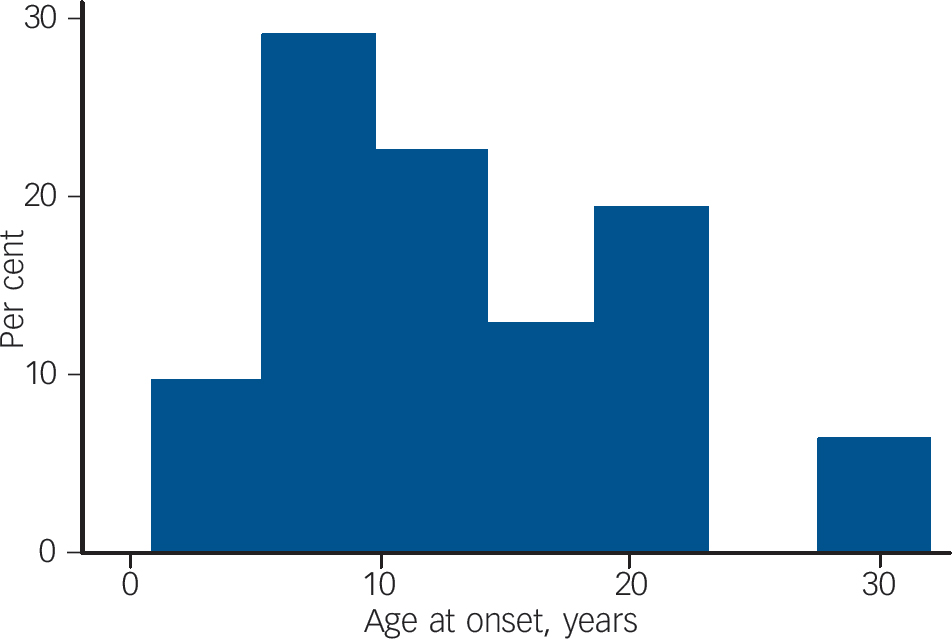
Fig. 1 Age at onset of seizures.
Frequency and severity of seizures
More than half (19) of the individuals with epilepsy had seizures occurring at weekly or less frequency (when the epilepsy was at its worst). Seizures occurred several times a day in just three individuals. However, these individuals also had seizure-free periods. Anticonvulsant drugs were being prescribed on an on-going basis for 29 participants. The majority of them (n = 26) were receiving one or two anticonvulsants. The remainder was being treated with more than three or more anti-epileptic drugs and information about medication was not available for two participants. Two individuals had not been prescribed medication for their epilepsy.
Age at onset of epilepsy
The mean age at onset of confirmed epilepsy was 13.3 years (s.d. = 7.8). The frequency histogram of age at onset of seizures is shown inFig. 1. This reveals that for the majority of participants, seizures first began after the age of 10 years. We compared the age at onset of epilepsy in our sample with the age at onset reported in other studies. Reference Kurtz, Tookey and Ross8,Reference Goulden, Shinnar, Koller, Katz and Richardson9 In the 1958 child development study, 124 children developed epilepsy and 47 (38%) developed seizures for the first time after the age of 10. Reference Kurtz, Tookey and Ross8 This proportion is significantly lower than the proportion observed (58%) in our study (binomial test P = 0.02) Likewise, in a Scottish cohort of 152 children with idiopathic mental retardation born between 1951 and 1955 and followed to age 22 years, Reference Goulden, Shinnar, Koller, Katz and Richardson9 37.5% developed epilepsy (n = 3/8) after the age of 10 years. Again this proportion is significantly lower than the proportion observed in our sample (binomial test P = 0.016).
Correlates of epilepsy
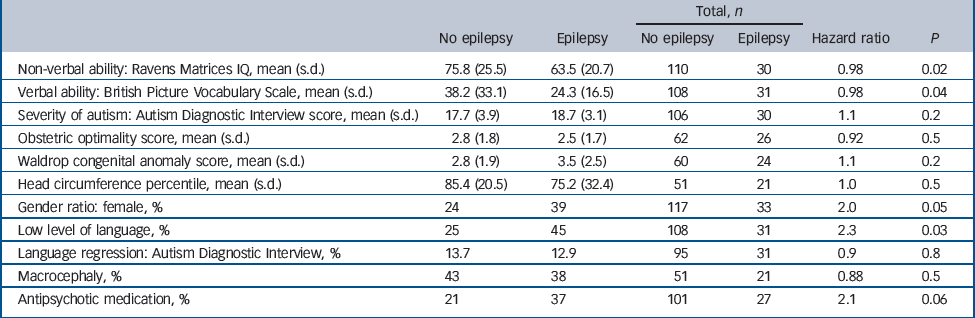
Table 2 summarises the characteristics of the individuals with and without definite epilepsy. In total, 18% (19/104) of male participants developed epilepsy compared with 30% (14/46) of the female participants and females were significantly more likely to develop epilepsy (hazard ratio (HR) = 1.99, s.e. = 0.71, P = 0.05). However, the age at onset of seizures in males (mean 12.2, s.d. = 1.7) was non-significantly (P = 0.2) higher than the age at onset in females (mean 8.9, s.d. = 1.7); similarly, males and females with epilepsy did not differ in their level of intellectual or language abilities.
Table 2 Proband features according to the presence of epilepsy
| Total, n | ||||||
|---|---|---|---|---|---|---|
| No epilepsy | Epilepsy | No epilepsy | Epilepsy | Hazard ratio | P | |
| Non-verbal ability: Ravens Matrices IQ, mean (s.d.) | 75.8 (25.5) | 63.5 (20.7) | 110 | 30 | 0.98 | 0.02 |
| Verbal ability: British Picture Vocabulary Scale, mean (s.d.) | 38.2 (33.1) | 24.3 (16.5) | 108 | 31 | 0.98 | 0.04 |
| Severity of autism: Autism Diagnostic Interview score, mean (s.d.) | 17.7 (3.9) | 18.7 (3.1) | 106 | 30 | 1.1 | 0.2 |
| Obstetric optimality score, mean (s.d.) | 2.8 (1.8) | 2.5 (1.7) | 62 | 26 | 0.92 | 0.5 |
| Waldrop congenital anomaly score, mean (s.d.) | 2.8 (1.9) | 3.5 (2.5) | 60 | 24 | 1.1 | 0.2 |
| Head circumference percentile, mean (s.d.) | 85.4 (20.5) | 75.2 (32.4) | 51 | 21 | 1.0 | 0.5 |
| Gender ratio: female, % | 24 | 39 | 117 | 33 | 2.0 | 0.05 |
| Low level of language, % | 25 | 45 | 108 | 31 | 2.3 | 0.03 |
| Language regression: Autism Diagnostic Interview, % | 13.7 | 12.9 | 95 | 31 | 0.9 | 0.8 |
| Macrocephaly, % | 43 | 38 | 51 | 21 | 0.88 | 0.5 |
| Antipsychotic medication, % | 21 | 37 | 101 | 27 | 2.1 | 0.06 |
Epilepsy was significantly more common in those individuals with very limited overall level of language (25% of those without epilepsy and 45% of those with epilepsy had a very limited overall level of language: HR = 2.3, P = 0.03). Individuals with epilepsy also had significantly lower verbal ability measured using the BPVS (HR = 0.98, s.e. = 0.007, P = 0.04) and non-verbal IQ measured with the Ravens Matrices (HR = 0.98, s.e. = 0.008, P = 0.02).
Cox's multiple regression analysis of the 137 participants with complete data showed that among these predictors, both non-verbal ability (HR = 0.98, P = 0.02) and proband gender (HR = 2.2, P = 0.03) remained significantly and independently associated with seizure outcome.
Medication
Review of the clinical notes and SAPPA interviews provided information about the prescription of psychotropic medication for 128 individuals. In total, 31 participants were reported to have taken psychotropic medication that might lower the seizure threshold and thus induce seizures. All of these individuals had taken antipsychotic medication at some time and ten had a history of epilepsy. Fourteen participants had been treated with antidepressants and five of these had a history of epilepsy. Four participants with epilepsy and nine without had taken both antidepressants and antipsychotics. There was a tendency (P = 0.06) for medication to be prescribed more often in females (12/31) than in males (20/97). There was also a tendency for the prescription of antipsychotic medication to be associated with a history of epilepsy (HR = 2.1, P = 0.06).
New medical conditions
For the large majority of individuals, special investigations (for example magnetic resonance imaging or computed tomography scan) to exclude the possibility of an undiagnosed medical condition (such as brain tumour) were not requested by the clinicians involved in their care. However, for individuals with severe, more intractable seizures, special investigations were sometimes undertaken. No new medical conditions that might lead to epilepsy were reported.
Familial liability
We first examined the family history data to compare the frequency of reported epilepsy in the first-, second- and third-degree relatives of our autism families with the rate of epilepsy in the relatives of a group of Down syndrome families. This comparison showed that 12/1613 (0.74%) autism relatives and 4/734 (0.54%) of Down syndrome relatives were reported to have epilepsy. These frequencies were not significantly different.
We also compared the frequency of epilepsy in the relatives from the autism families according to the presence of epilepsy in the autistic proband. This showed that 8/1057 (0.76%) of relatives of probands without epilepsy had a history of seizures compared with 3/497 (0.6%) of relatives of probands with epilepsy. Again this difference was not significant. These findings indicated that epilepsy was neither more common in autism families nor in relatives of probands with epilepsy compared with those without.
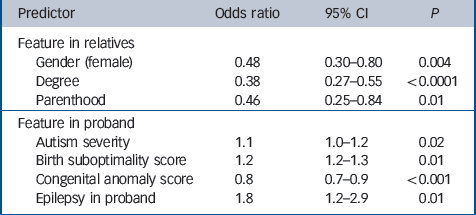
Next we examined the relationship between the presence of epilepsy in the autistic proband and the likelihood of a relative (first-, second- and third-degree) exhibiting the broader autism phenotype. Our previous research has shown the liability to the familiality of autism was associated with various characteristics of the proband (severity of autism, number of minor congenital anomalies, obstetric optimality score) and the relative (gender, genetic relationship to the proband and whether they were ascertained for parenthood). These analyses were undertaken using logistic regression with robust variance estimates. The findings are summarised inTable 3 and indicated that the presence of epilepsy in the proband was a significant predictor of the risk of a relative having the broader autism phenotype even in the presence of the other predictors. The association between epilepsy in the proband and the likelihood of a relative having the broader autism phenotype was not dependent on whether the family history was obtained before or after the onset of seizures in the proband or on the social class or educational status of the informant parent (data not shown). Thus, there was no evidence of any systematic reporting bias that might account for the association.
Table 3 Predictors of the broader phenotype in relatives (1295 relatives)
| Predictor | Odds ratio | 95% CI | P |
|---|---|---|---|
| Feature in relatives | |||
| Gender (female) | 0.48 | 0.30–0.80 | 0.004 |
| Degree | 0.38 | 0.27–0.55 | <0.0001 |
| Parenthood | 0.46 | 0.25–0.84 | 0.01 |
| Feature in proband | |||
| Autism severity | 1.1 | 1.0–1.2 | 0.02 |
| Birth suboptimality score | 1.2 | 1.2–1.3 | 0.01 |
| Congenital anomaly score | 0.8 | 0.7–0.9 | <0.001 |
| Epilepsy in proband | 1.8 | 1.2–2.9 | 0.01 |
Although the gender of the proband was not associated with the likelihood of a relative having the broader phenotype when it was entered into the regression model, there was some evidence for an interaction between proband gender and epilepsy, with the model ‘fit’ significantly improving when the interaction term was included in the model (change in Wald χ2 = 10.36, d.f. = 1). However, with the interaction term included in the model, the odds ratio for proband gender, epilepsy and the interaction term all became non-significant.
Discussion
Strengths and limitations
The strengths of this study lie in the long-term follow-up of a well-characterised sample of individuals with autism and the availability of good-quality data on the probands and their relatives. There are also several limitations. First, much of the information was retrospectively gathered and based on parent report, so may have been subject to recall and other biases. Second, it was not possible to systematically undertake EEG studies, which may have aided in classification and the identification of potential epileptic foci. Third, the study sample was selected from a clinic population following the exclusion of individuals with very severe or profound intellectual disabilities and those with infantile spasms. In addition, for the purpose of the family study, participants were stratified by gender and IQ group. As such, the results reported here apply to a subset of individuals with autism and the effect sizes will differ to those derived from population-based studies. The exclusion of individuals who were very severely and profoundly intellectually disabled is likely to have influenced our estimate of the prevalence of epilepsy in individuals with autism, as will have the exclusion of participants with infantile spasms.
Despite the limitations, the individuals we have studied are fairly typical of those seen and diagnosed in child psychiatry. Moreover, the follow-up into adulthood provides special insights into the pattern of seizures that arise (given that seizures often begin after middle childhood).
Main findings
The key findings from the study were that:
-
(a) epilepsy developed in around 22% of individuals;
-
(b) seizures began after the age of 10 in the majority of the sample and did not start until adulthood in some;
-
(c) the distribution of age at seizure onset is significantly different to the distribution reported in the general population in the UK Reference Kurtz, Tookey and Ross8 as well as that reported in individuals with intellectual disability. Reference Goulden, Shinnar, Koller, Katz and Richardson9 The different pattern is unlikely to be a result of sampling bias, as seizures in our cohort usually began after the children were enrolled in the sample. It is possible, however, that the exclusion of individuals with severe and profound intellectual disability may have influenced the pattern observed in the age at onset of seizures.
The comparatively late age at onset of epilepsy means that in the vast majority of individuals, epilepsy usually develops well after the emergence of the behavioural syndrome becomes evident. This is clearly incompatible with the notion that clinical seizures caused autism in our participants, Reference Tuchman and Rapin5 although it leaves open the possibility that early-onset seizure disorders may be causally relevant in the comparatively rare situation where seizures begin in infancy. Reference Saemundsen, Ludvigsson, Hilmarsdottir and Rafnsson26
The seizure disorder in this sample was usually characterised by generalised tonic–clonic seizures. Simple and complex partial seizures also occurred, but only in the context of a concomitant generalised tonic–clonic seizure disorder. The profile of seizure types found in this study differs from that observed in other reports, where partial seizures were more common. It is possible that the generalised tonic–clonic seizures described actually represented secondary generalised seizures arsing from an epileptic focus, but more detailed studies would be required to test this.
Except for a few participants, the EEG investigations undertaken at the time revealed non-specific abnormalities that did not aid much in classification or clinical management. However, most of the EEG studies were conducted many years ago and modern day investigation may prove more clinically fruitful. It was also notable that seizures occurred on a weekly basis or less in the majority of individuals, and could often be controlled after instigating just one or two anti-epileptic drugs.
None of the individuals studied in this sample was reported to have developed a new medical condition that might explain the development of late-onset seizures. It appears, therefore, that when seizures develop in young people and adults with autism, this does not ordinarily signify the presence of a new medical condition, such as a brain tumour, as it may do in individuals without autism. This provides some justification to the practice of only undertaking special investigations in the light of the full clinical picture.
In our study, a history of epilepsy tended to be associated with the prescription of antipsychotic medications that may lower seizure threshold. The tendency approached statistical significance, but the basis for it is unclear for several reasons. To begin with, the information about the prescription of medications was limited in terms of how systematically it was collected. Thus, we asked systematically about the use of medication among individuals who had significant changes in behaviour and those who had epilepsy. This was supplemented with information from the medical records, but even so, we did not have systematic data on everyone. As such, measurement error or bias cannot be ruled out. In addition, we lacked details concerning the time points when medications were prescribed. Therefore, the temporal relationship with seizure onset remains unclear, so rather than potentially precipitating the onset of seizures, it is possible that medications were being prescribed to treat disturbed behaviour in people who had already developed epilepsy. The findings are consistent with those reported in another study and highlight the need for more research on this topic, Reference Hara12 especially as the use of medication in treatment of autism-spectrum disorders is increasing. Reference Aman, Lam and Van Bourgondien27 Clinicians need to be aware that individuals with autism are prone to developing late-onset seizures and therefore that drugs that might lower seizure threshold should be prescribed with caution.
The likelihood of developing epilepsy was increased in females and those with lower verbal and non-verbal abilities. Both the level of ability and gender of the proband were independently associated with the liability to seizures. It was noteworthy, however, that several key associated features (head size/the presence of macrocephaly, the optimality of the pregnancy and birth and the number of minor congenital anomalies) were not correlated with the risk for epilepsy. The power to detect associations was limited, but our findings are consistent with the results of previous reports in showing that epilepsy in autism is associated with gender and ability level, but not other features of autism. Reference Amiet, Gourfinkel-An, Bouzamondo, Tordjman, Baulac and Lechat6,Reference Parmeggiani, Posar, Giovanardi-Rossi, Andermann and Zifkin28,Reference Baird, Charman, Pickles, Chandler, Loucas and Meldrum29 The reason for this pattern of findings is uncertain, but may point to aetiological heterogeneity. Individuals with autism and severe/profound intellectual disability may be more aetiologically heterogeneous, but were not included in our study,
We found that individuals with epilepsy had lower intellectual, speech and language abilities than those without epilepsy, but epilepsy was not associated with a history of autistic regression. The association with lower ability has been noted in a number of studies. Reference Bartak and Rutter30 However, the findings with regard to regression have been inconsistent, with some studies finding an association Reference Hrdlicka, Komarek, Propper, Kulisek, Zumrova and Faladova31 and others not. Reference Baird, Charman, Pickles, Chandler, Loucas and Meldrum29,Reference Spence and Schneider32 Differences in sampling and the variable period of follow-up, as well as the retrospective nature of most investigations may account for at least some of these differences, but at present an association between epilepsy and regression in autism has not been firmly established.
We also showed that the relatives of people with autism were no more likely to develop epilepsy than the relatives of children with Down syndrome. Moreover, the presence of epilepsy in the probands with autism was not associated with an increased risk of epilepsy among their relatives. This picture contrasts to that observed in the relatives of individuals with idiopathic generalised epilepsy, where the recurrence risk of epilepsy is increased four- to ninefold and the rate of epilepsy in first-degree relatives is 4–8%. Reference Helbig, Scheffer, Mulley and Berkovic33 The small number of participants with epilepsy in our sample preclude us from ruling out an increased risk of epilepsy among relatives, but if our results are confirmed they suggest that quite different mechanisms are operating in autism.
The presence of epilepsy in the proband was associated with the liability to the familiality of the broader phenotype of autism. Yet, relatives with the broader autism phenotype were not at increased risk for epilepsy. These results are probably best understood within a liability threshold model of autism. This suggests that the genetic and environmental factors that underlie the liability to the familiality of autism also confer a risk for epilepsy. What familial factors might be involved? One possibility is that there is a shared genetic risk. Participants with an established causal genetic condition (for example tuberous sclerosis and fragile-X syndrome) were excluded from this study and although undetected genetic abnormalities may have been present in a few individuals the rarity of such abnormalities would make this an unlikely explanation for the findings. Moreover, single gene and genomic disorders cannot account for the association between the risk of epilepsy and the familial liability to the broader autism phenotype.
The fact that seizures often started in adolescence raises the possibility that the processes involved in remodelling the brain and pruning synapses may be relevant, especially as there is evidence that age-related changes in cortical thickness might differ in individuals with autism-spectrum disorder. Reference Raznahan, Toro, Daly, Robertson, Murphy and Deeley34 However, as yet, there are few clues concerning the mechanisms responsible for the increased risk for epilepsy and late age at onset of seizures in individuals with autism.
Funding
P.F.B. was supported by the National Institute of Health Research, Biomedical Research Centre in Mental Health. The study was supported by grants from the UK Medical Research Council.
Acknowledgements
We wish to thank the individuals and their families who participated in this research.





eLetters
No eLetters have been published for this article.