


Optimal environment is crucial for child development and brain health across the lifespan. Positive environmental conditions such as nutrition and social support, as well as negative conditions such as stress, inflammation and toxin exposure all contribute to the integrity of developing organs, including the brain(Reference Wachs, Georgieff and Cusick1). When environmental perturbations take place early in life, e.g. the first 1000 d from conception, the environmental effects can have long-lasting or lifespan consequences.
The role of early nutrition in shaping life-time health outcomes became prominent in the 1990s with the work of David Barker and others who noted that an adult's risk of cardio-metabolic disease was linked to intrauterine nutritional status and birth weight(Reference Gluckman and Hanson2). Subsequently, nutrition's role in the first 1000 d has been explored with respect to relevant outcomes beyond cardiovascular/metabolic health, including immune integrity, bone health, risk of cancer and, most recently brain (mental) health(Reference Raiten, Steiber and Carlson3).
The interest in early nutrition and brain development has been fostered through some remarkable breakthroughs in mechanistic understanding of how nutrients interact with the developing brain and led to new thinking about delivery and assimilation of nutrients. These insights, in turn, have opened new horizons for therapeutic interventions, including nutritional and non-nutritional solutions to nutrient perturbations of deficiency or excess. However, challenges remain in the realm of proving conclusively that a specific nutritional condition or intervention actually affects brain structure and function, particularly at the individual level.
This chapter discusses some of the remarkable breakthroughs, new horizons and challenges in the field of early life nutrient–brain interactions by using what has been discovered in the field of iron and brain biology as a paradigm for how these interactions occur and are regulated.
Breakthroughs
Two recent breakthroughs in scientific information and conceptualisation have significantly affected how scientists are currently considering nutrient–brain interactions: the developmental origins of health and disease concept applies to the brain and the discovery of potential mechanisms underlying the long-term mental health effects of early nutrition. The lessons learned from these breakthroughs are beginning to inform therapeutic approaches to nutritional perturbations that affect the brain.
The developmental origins of health and disease concept applies to the brain
The current developmental origins of health and disease concept is an outgrowth and refinement of the Barker hypothesis as it was refined by Gluckman and Hanson in the early 2000s(Reference Gluckman and Hanson2). Clear mechanistic evidence across multiple health domains indicates that the first 1000 d post-conception influence child and adult health outcomes. The idea is not new – the poet William Wordsworth codified the concept in literature in 1802 when he wrote that the child is the father of the man. Nevertheless, it was Barker's original studies linking the risk of adult-onset diabetes mellitus, hypertension and CVD to intrauterine growth restriction that brought the field into the modern era with two key mechanistic findings. First, mechanistic studies implicated the roles for pro-inflammatory cytokines and the hypothalamic–pituitary–adrenal axis hormones, including activation of cortisol in altering the regulation of metabolic setpoints for glucose and insulin regulation(Reference Howland, Sandman and Glynn4). Secondly, an important conceptual breakthrough revealed that fetal and early postnatal life is a time when the regulation of certain genes was set for the lifespan(Reference Joss-Moore and Lane5). While initially applied to cardiovascular health, both factors proved important for brain health as well(Reference O'Donnell and Meaney6).
Optimal brain development is important for its current functional capacity, but also sets the stage for later developmental stages and function(Reference Wachs, Georgieff and Cusick1). The early years from conception to approximately 3 years postnatal age see the brain grow from a smooth, bilobed structure at 5 months gestation to a complex sulcated and gyrified structure by full-term birth. It is important to understand that the brain is not a homogenous entity and should not really be conceptualised as a single structure. Instead, it is useful to think of it as multiple regions and processes, each with its own developmental trajectory(Reference Thompson and Nelson7). The early years encompass the development of primary systems such as the hippocampus/striatum, myelination and neurotransmitters, which in turn support primary neurobehavioural processes such as learning and memory formation, speed of processing and reward mechanisms, respectively. These systems are at greatest risk for nutrient perturbations during their period of most rapid growth(Reference Wachs, Georgieff and Cusick1). Systems that develop later in childhood, such as the prefrontal cortex, are highly dependent on the integrity of these early developing ones. The frontal lobe receives projections from the earlier developing hippocampus and striatum and relies on the integrity of those structures as well as the efficiency of processing via myelination and neurotransmitters to ensure proper connectivity. Thus, each stage of brain development depends on the previous stage, essentially creating a scaffolding effect(Reference Wachs, Georgieff and Cusick1). Nutrient perturbations that disturb the integrity of the primary structures have ‘ripple effects’ on the later developing structures even though the nutrient perturbation might not have directly damaged it. It is not unusual to see late effects of nutrient intervention studies on frontal lobe functions (e.g. attention, planning) that were not obvious at earlier assessment timepoints(Reference Jabès, Thomas and Langworthy8,Reference Colombo, Shaddy and Gustafson9) .
The young brain is remarkably dynamic in its growth and development and exhibits a great deal of plasticity. Brain growth is a highly metabolic process. Sixty per cent of a baby's energy expenditure is devoted to the brain, compared with only 20 % in the adult(Reference Kuzawa10). Moreover, the baby's oxygen consumption rate is nearly double that of the older child. Nutrients that support oxidative metabolism and ATP generation by mitochondria must be present in adequate supply to support this metabolic demand of rapid regional brain growth, which includes not only neurons but glial cells as well. Nutrients that particularly support this structural/functional development include oxygen, glucose, amino acids, iron, copper, iodine, folate, vitamin B12 and choline.
All nutrients are important for all cells to function, but certain nutrients and nutritional conditions particularly affect early brain development (Table 1). Perturbation of these nutrients can cause acute brain dysfunction during the period of deficit or excess. The long-term effects on adult function are of greatest concern because they result in the true cost to society of unfulfilled educational or job potential and the burden of mental health disorders(Reference Walker, Wachs and Gardner11). Each of the nutrients noted in Table 1 exhibits a critical or sensitive period for regional neurodevelopment. Some alter regulation of synaptic plasticity genes and functional gene networks through their ability to alter the epigenetic landscape as discussed later (Table 2).
Table 1. Nutrients and nutritional conditions that affect early brain development in pre-clinical models and/or human subjects
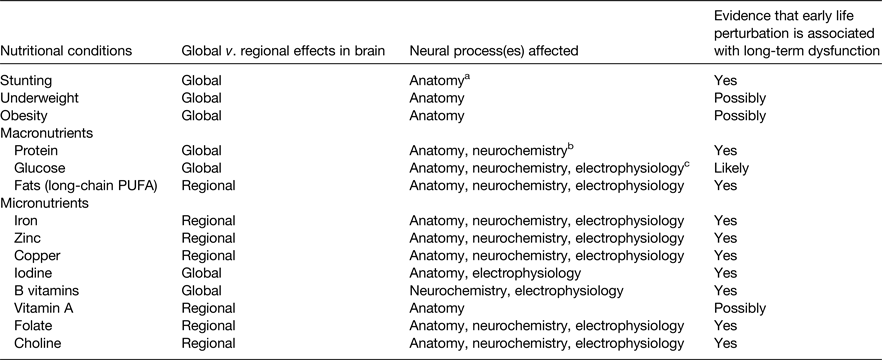
a Neuroanatomy includes neurons and glial cells.
b Neurochemistry includes synthesis of neurotransmitters.
c Neurophysiology includes propagation of electrical impulse.
Table 2. Nutrients and nutritional conditions that induce epigenetic modifications to chromatin in the fetal and neonatal brain

Early life iron deficiency illustrates the principles of how a nutrient can confer acute and long-term risks to brain function. Iron deficiency at birth compromises the integrity of the hippocampus and results in poorer neonatal recognition memory function(Reference Siddappa, Georgieff and Wewerka12,Reference Geng, Mai and Zhan13) , slower speed of neural processing(Reference Amin, Orlando and Eddins14) and poorer bonding and maternal interaction(Reference Wachs, Pollitt and Cueto15). The hippocampal effects remain present (as a function of neonatal iron stores) until at least 4 years of age(Reference Riggins, Miller and Bauer16), thus demonstrating long-term effects long after iron deficiency has resolved.
Timing of a nutrient deficiency matters. Fetal iron deficiency is associated with a higher risk of autism, schizophrenia and neurocognitive dysfunction dependent on whether the deficiency occurred in the first, second or third trimester, respectively(Reference Schmidt, Tancredi and Krakowiak17–Reference Tamura, Goldenberg and Hou19). Lower iron stores in the newborn also leads to earlier onset of postnatal iron deficiency in infancy and toddlerhood with its attendant long-term sequelae of motor dysfunction, socio-affective disorders and increased risk of depression and anxiety in adulthood(Reference Shafir, Angulo-Barroso and Jing20–Reference Lukowski, Koss and Burden22). Given the high prevalence of iron deficiency worldwide, prevention of the long-term effects would have a large medical and economic effect on the population.
Potential mechanisms underlying the long-term effects of early nutrition on brain function
A second set of major breakthroughs occurred as researchers discovered mechanisms underlying the long-term effects of early nutrient disorders. These breakthroughs have provided useful clinical information with respect to prevention, screening, repletion and unique therapies related to early life nutritional disorders. Once again, iron deficiency can function as a paradigm for mechanisms by which nutrients affect long-term brain function.
Two major theories account for the loss of long-term neurodevelopmental potential, instantiated by the loss of synaptic plasticity and brain performance: (1) critical periods of brain development and (2) epigenetic modification of synaptic plasticity genes/gene networks. The two theories are not mutually exclusive and indeed likely interact with each other.
Critical periods
The first theory depends on the principles of critical periods of development(Reference Hensch23). The young brain has limited performance ability and efficiency. However, it is highly plastic and resilient to stressors. Its ability to repair itself following injury or to respond to treatment exceeds that at any other time in life. Brain development is not uniform. Instead, the various brain regions and cell types have unique developmental trajectories, with different start and end times(Reference Thompson and Nelson7). Each region has an epoch during which development progresses more rapidly, referred to as its sensitive or critical period. Some regions, e.g. cerebellum, have a broad trajectory of development, while other areas are temporally more highly focused, e.g. hippocampus. The nutritional requirements during the critical period of rapid development are greater than during periods of slower rates of development(Reference Kuzawa10). A perturbation of nutrient status, such as iron deficiency, during the critical period can result in structural and functional changes that persist into adulthood(Reference Jorgenson, Wobken and Georgieff24–Reference Pisansky, Wickham and Su27). The neurobehavioral deficits in adulthood relate directly to the disordered neuronal structure(Reference Jorgenson, Wobken and Georgieff24,Reference Jorgenson, Sun and O'Connor25,Reference Pisansky, Wickham and Su27) . Once a region has progressed through its critical period, it exhibits much greater efficiency but concomitantly loses its plasticity, making it less responsive to treatments such as nutrient repletion(Reference Hensch23,Reference Callahan, Thibert and Wobken28) and persistence of neurobehavioral and structural findings long after the nutrient deficiency has resolved.
The hippocampus is a highly metabolic brain region that develops rapidly starting in the last trimester at 28 weeks gestation until approximately 2 years of postnatal age in human subjects. The developmentally equivalent time frame in the rodent is postnatal day 5 to postnatal day 25. Since highly metabolic cells require nutrients that support oxidative metabolism to generate ATP, it is reasonable to ask whether there is a critical period for one such nutrient, iron, during hippocampal development. A major breakthrough in iron–brain nutrition occurred with the generation of a non-anaemic mouse model of specific hippocampal neuronal iron deficiency. The deficiency was timed to occur during the period of rapid hippocampal growth(Reference Fretham, Carlson and Wobken26). Iron uptake by hippocampal CA1 neurons depends on the cell membrane surface iron transporter, transferrin receptor-1. A non-functional dominant-negative transferrin receptor was genetically introduced specifically in the hippocampus of mice just prior to the onset of the putative critical period. The functional wild-type receptor could be reintroduced at any time by the addition of doxycycline to the animal's diet. The investigators reintroduced the wild-type receptor either during the putative critical period (postnatal day 21) or afterwards (postnatal day 42)(Reference Fretham, Carlson and Wobken26). Treatment during the critical period at postnatal day 21 rescued the structural and behavioural abnormalities induced by neuronal iron deficiency, while treatment after the critical period did not. This was the first clear demonstration that iron was essential to the developing neuron and iron deficiency itself, without anaemia, is a causative factor for the long-term structural and behavioural abnormalities, thus implicating a critical period for iron in hippocampal development(Reference Fretham, Carlson and Wobken26). While this example is quite specific with respect to nutrient (iron) and brain region (hippocampus), the principles apply to iron and other brain regions (e.g. frontal cortex, cerebellum), processes (e.g. myelination, neurotransmitters). Furthermore, other nutrients including iodine, zinc, glucose, protein, copper, choline and vitamin B12 influence neuronal structure, myelination and neurotransmitter during development and thus the principles likely apply to them as well.
Epigenetic modification
The second theory regarding the long-term effects of early life nutrition depends on emerging knowledge that certain, but not all, nutrients have been shown to alter gene expression through epigenetic modification of chromatin in the brain (Table 2). Epigenetic modification of chromatin occurs in multiple ways to either close off or open up the DNA for transcription. The modifications can be temporary or long-term/permanent thereby altering the activity of the gene and the protein it is coded to produce. While a description of all epigenetic processes is well beyond the scope of this article, the major ways in which the nutrients listed in Table 2 can affect genes in the brain include Cytosine-phosphate-Guanine (CpG) methylation and hydroxymethylation, and histone methylation and acetylation.
For some nutrients (e.g. iron), the direct role of the nutrient in the process of certain types of epigenetic modification is understood. The iron–nickel pocket of the class of histone demethylases called Jumonji- and AT-rich interaction domain-containing proteins regulates the rate of histone methylation, which in turn regulates the rate of transcription(Reference Sengoku and Yokoyama29). Iron deficiency alters the binding affinity of the Jumonji- and AT-rich interaction domain-containing protein particularly through gene activation marks at lysine residue (K) 4 and gene repressive marks at K27 in the promoter region of a key synaptic plasticity gene, brain-derived neurotrophic factor (BDNF)(Reference Tran, Kennedy and Lien30). In a rodent model of gestational/lactational iron deficiency, the enrichment of the repressive mark (K27) is increased and the activation mark (K4) is decreased in the BDNF-4 gene in the hippocampus both at postnatal day 15 during the period of acute iron deficiency as well in young adulthood, long after the iron deficiency has resolved. BDNF mRNA and protein expression are both reduced as a result and the animal behaviourally shows poorer hippocampal as evidenced by worse learning and memory performance(Reference Tran, Kennedy and Lien30). Germane to this discussion, the epigenetic marks remain in place during adulthood even though the hippocampus is iron sufficient by that point(Reference Tran, Kennedy and Lien30).
For other nutrients, a deficiency can induce demonstrable effects on the epigenetics of the brain that are not yet understood mechanistically based on the biology of the nutrient. These could represent secondary effects, e.g. the deficiency causes stress which in turn alters brain DNA methylation patterns(Reference O'Donnell and Meaney6).
The list of nutrients identified to cause changes to DNA methylation in early life has been growing. These early life nutrients not only alter the expression of salient and specific genes in the brain such as BDNF across the lifespan, but also alter the expression of gene networks related to brain function and neuropathologic processes. The use of genome-wide screening tools such as RNA-seq has elucidated these effects and the altered genes have been grouped through informatic approaches into those identified pathways(Reference Tran, Kennedy and Pisansky31). Early life iron deficiency can serve as a paradigm for nutrient–brain pathway interactions involving gene networks. A rat model of gestational iron deficiency followed by neonatal iron treatment and repletion demonstrated changes to networks of genes coding for autism, schizophrenia and Alzheimer's disease(Reference Tran, Kennedy and Pisansky31). The first two are of particular interest because they directly confirm the epidemiologic findings in human subjects of an increased risk of those diseases following gestational iron deficiency(Reference Schmidt, Tancredi and Krakowiak17,Reference Insel, Schaefer and McKeague18) .
New horizons
Breakthroughs in the understanding of the neurobiology of nutrient–brain interactions can lead to new horizons for prevention and therapy. One example involves leveraging the knowledge of when critical periods for nutrients occur in order to prevent deficiencies or to initiate treatments in a more timely manner. A more transformative example involves designing nutritional workarounds to mitigate the long-term effects of early nutrient deficiencies, particularly those mediated by epigenetic modifications. The strategy of more timely treatment seems relatively obvious, but implementation of such strategies is hampered by lack of knowledge of when critical or sensitive periods for any given nutrient occur in various brain regions and the lack of biomarkers/bioindicators that read out the nutrient status in the brain and its effect on brain function (see next section)(Reference Tran, Kennedy and Pisansky31).
Conversely, exciting new data indicate that epigenetic modifications induced by nutrient deficiencies can be treated with dietary components such as choline that have the capacity to reshape the epigenetic landscape to the positive. Many of the epigenetic effects documented with nutrients revolve around methylation of either CpG islands or histones (Table 2). These may be amenable to time-targeted therapy with choline, or other potential methyl donors(Reference Zeisel32). Timed prenatal and early postnatal choline has been shown to improve hippocampal outcomes in normal rats(Reference Meck, Smith and Williams33), rats with fetal alcohol exposure(Reference Ryan, Williams and Thomas34), the Down syndrome mouse(Reference Moon, Chen and Gandhy35) and mice with the genetic mutation that causes Rett's syndrome(Reference Ricceri, De Filippis and Laviola36). The fact that multiple species with disparate neurologic risk factors including genetic abnormalities and toxin exposure all respond to the treatment suggests that a fundamental biological mechanism is being addressed(Reference Zeisel32).
Recently, our group showed that prenatal choline delivered to iron-deficient gestating rat dams partially reverses the suppression of BDNF mRNA and protein expression in the hippocampus of their offspring accompanied by re-activation of the K4me3 epigenetic mark and re-suppression of the inhibitory K27 mark(Reference Tran, Kennedy and Lien30). We found that hippocampal recognition memory behaviour (as indexed by preferential looking time) in the offspring, which was compromised by gestational iron deficiency, was restored by the choline treatment either in the prenatal or early postnatal period(Reference Kennedy, Dimova and Siddappa37,Reference Kennedy, Tran and Kohli38) . Most importantly, the rescued epigenetic, gene regulatory, protein and behavioural findings remained present in the offspring in adulthood suggesting that such therapies have sustained and potentially permanent effects(Reference Tran, Kennedy and Lien30,Reference Tran, Kennedy and Pisansky31,Reference Kennedy, Dimova and Siddappa37,Reference Kennedy, Tran and Kohli38) .
The effects on single genes that are important for synaptic plasticity such as BDNF are exciting but genes do not typically work in isolation to mediate neuronal activity and behaviour. Gene networks drive neurologic functions and are also targeted in neurobehavioral disorders. To that end, we utilised RNA-seq methodology to study the effect of prenatal choline given to iron-deficient pregnant dams on gene networks in the hippocampus of the offspring in adulthood. Whereas gestational iron deficiency had activated gene networks implicated in schizophrenia, autism and Alzheimer's disease, gene networks implicated in autism, mood disorders and pervasive developmental disorders were rescued by choline despite the presence of iron deficiency(Reference Tran, Kennedy and Pisansky31). Trials of methyl donor diets in other nutrient deficiency states that induce epigenetic changes in the brain (Table 2) have not been frequently performed, but such trials present a golden opportunity for testing therapeutic approaches that could correct aberrant developmental trajectory courses.
The responses to pre- or postnatal choline supplementation in iron deficiency models (or for that matter any other nutrient deficiency model) have not yet been translated directly into human subjects. Clinical trials of choline for iron deficiency remain on the horizon. However, clinical trials of pre- and postnatal choline for children with fetal alcohol spectrum disorder(Reference Jacobson, Carter and Molteno39–Reference Ernst, Gimbel and de Water41) demonstrate the feasibility of translation from the preclinical models and the effectiveness of the therapy in human subjects. Prenatal supplementation of heavy drinking mothers with choline improved brain volumes and cognitive performance(Reference Warton, Molteno and Warton42). An ongoing trial of a 6-month choline v. placebo treatment course for children aged 2–5 years with fetal alcohol spectrum disorder shows improved hippocampal and prefrontal cortex-mediated behaviours up to 4 years post-treatment(Reference Wozniak, Fink and Fuglestad40). The research on the positive effect of postnatal choline supplementation in children with fetal alcohol spectrum disorder is particularly intriguing with respect to iron deficiency because fetal alcohol exposure impairs iron transport into the brain, rendering the brain iron deficient(Reference Warton, Molteno and Warton42,Reference Miller, Roskams and Connor43) . Direct evidence that these effects are due to epigenetic landscape changes is not being tested in the ongoing trial and thus one can only hypothesise that the effects are due to those documented in the preclinical model.
The new horizon of utilising ‘nutritional workarounds’ to correct or prevent the brain effects of nutritional deficits during development is particularly exciting for populations that may have restricted access to nutrients that are critical for brain growth and development. Once again iron can serve as an example. Iron deficiency is the most common micronutrient deficit in the world affecting close to 2 billion people, mostly women and children. The most bioavailable source of iron is meat, which is economically out of reach for many populations. Screening for iron deficiency, if performed at all, is done by assessment for the presence of anaemia. Yet, research in all young mammals including human subjects shows that the brain tissue becomes iron deficient before anaemia is present, thus effectively rendering the brain at long-term neurodevelopmental risk prior to diagnosis (see next section)(Reference Georgieff44). Universal iron supplementation is potentially dangerous in malaria endemic areas because the parasite that causes the disease thrives on iron found in erythrocytes; essentially iron deficiency anaemia is protective from severe malarial disease. If time-targeted choline treatment can reverse some, if not all, of the untoward neurologic effects of early life iron deficiency, particularly without additional iron supplementation, these populations may be well served. Recent work in cell culture models of iron deficiency in developing hippocampal neurons suggests that choline restores dendritic structure and neuronal metabolism compromised by iron deficiency even without iron treatment(Reference Bastian, von Hohenberg and Kaus45).
A significant challenge in defining nutrient–brain interactions
As noted in previous sections, a significant limitation in the field of nutrient–brain interactions is assessing a nutrient's status in the brain by assessing peripheral blood biomarkers. Each nutrient has a hierarchy of distribution within the body and without direct access to the brain, it is unclear which organs' nutrient status a biomarker measured in the peripheral blood is indexing. This is especially problematic for detection of brain status because that organ is insulated by the blood–brain barrier, often with active transport occurring at that level. Thus, the question is: How do we know whether a change in nutrient status affects the human brain?
The burden of proof of nutrient–brain interactions has relied on the integration of evidence across multiple levels of assessment in pre-clinical models and linking them in a developmentally appropriate manner to levels of evidence available in human research. Ultimately, the outcome of interest is neurobehavioral function in human subjects since behaviour is the efferent expression of the brain's activity. Human behaviours and especially psychopathologic behaviours are functions of complex circuitry that may not be reproducible in pre-clinical animal models. Animal models of autism and schizophrenia rarely recapitulate the entire clinical syndrome as seen in human subjects. Conversely, certain primary fundamental behaviours such as motor responses and recognition memory are remarkably similar in common pre-clinical models such as rats, mice, pigs and non-human primates as they are in human subjects. Developmental trajectories of the brain structures (e.g. neuroanatomy, connectivity) that subserve these primary systems are known and the information about timing and duration can be leveraged in asking questions about nutrient effects during neurodevelopment. Nutrients that have significant roles in structural development include protein, energy, iron, copper, zinc, iodine, vitamin B12 and choline. Structure can be analysed by neuroimaging in human subjects and by neuroimaging and direct inspection (i.e. microscopy) in pre-clinical models in order to draw parallels.
However, anatomy is fundamentally inert. Behavioural function is driven by activating the anatomy through the neurochemistry and neurometabolism of the brain. Nutrients that affect the electrophysiology of the brain include glucose, protein, iron, zinc, copper, iodine, magnesium, calcium, phosphorus and common electrolytes among others. The brain's electrical performance can be assessed in human subjects via scalp surface electroencephalography, particularly using functionally linked techniques such as event-related potentials. Preclinical models can be assessed similarly, but also can be investigated via direct invasive in vivo and ex vivo neuronal recording. The biochemistry underlying the neurometabolism can be assessed in human subjects via magnetic resonance spectroscopy as it can be in preclinical models, where direct assessment can provide final proof. Understanding mechanisms through gene regulation is of importance. Pre-clinical models have shown the profound influences of nutrients on regulation of synaptic plasticity genes and gene networks during development. While gene expression is easily assessed in pre-clinical models because brain tissue is available, it has generally been precluded in human subjects because of the lack of brain tissue-based evidence.
Current approaches that assess nutrient effects do so by measuring biomarkers of the nutrient of interest. Typically, these biomarkers are measured in a fluid or tissue that is available and poses no or minimal risk in procurement from the subjects, often whole blood, plasma/serum or urine. All reside on the proximal side of the blood–brain barrier and may bear little or no relationship to the brain's nutrient or functional status. ‘Normal’ values are derived from population reference values usually from large data bases. When followed over time, available nutritional biomarkers can show trends in nutrient status. However, the relationship of values derived by population statistical cut-offs (e.g. ±2 standard deviations) to neurophysiologic consequences of the nutrient on the brain is loose, at best, and thus does not inform about the effect of the nutrient on brain biology or function. Few, if any, nutrients have a signature effect on brain function.
Identification and measurement of bioindicators as opposed to or in addition to nutritional biomarkers is an emerging field in nutritional neuroscience(Reference Combs, Trumbo and McKinley46). The goal of bioindicators is to assess whether a nutrient (or change in nutrient status) has affected the brain's function by obtaining readouts of biologically plausible perturbed physiology induced by the nutrient. Ideally, the metric would be easily accessible (e.g. serum, urine, saliva) but would reflect changes in brain that would be predictable based on the nutrient's known biological function.
For iron, detection of anaemia by measuring Hb concentration is currently used as the primary screening tool for iron deficiency. Yet, extensive research indicates that the young child's brain iron status is compromised before anaemia is present(Reference Georgieff44), which renders this easy to perform biomarker practically useless to prevent potential long-term brain dysfunction. In contrast, two forms of bioindicators that can be measured in the serum and leverage the known brain iron biology iron from preclinical models show promise in giving a more direct view of the effect of iron deficiency on the young brain: (1) measurement of the ratio of products of oxidative metabolism and (2) analysis of neurally derived exosomes.
One major effect of tissue-level iron deficiency is compromised oxidative metabolism and ATP generation(Reference Bastian, von Hohenberg and Georgieff47) with consequent effects on lactate and pyruvate generation. In a monkey model of spontaneous, progressive dietary infantile iron deficiency, like what occurs in human infants, targeted metabolomic approaches were used to simultaneously assess serum and cerebrospinal fluid. Parallel alterations to the citrate/pyruvate ratio in serum and spinal fluid were found 2 months before the onset of anaemia, indicating that brain metabolism at this timepoint was already compromised and that this compromise could be detected in the serum(Reference Sandri, Kim and Lubach48). This ratio could serve as an early warning of a brain at risk for the negative consequences of infantile iron deficiency.
Another exciting approach that has emerged utilises neurally derived exosomes from the brain that are found in the serum and analysed for protein or metabolites. Utilising knowledge of iron deficiency effects on brain protein and metabolic processes, one can assess whether changes have occurred in an individual from whom these exosomes have been sampled. The synaptic plasticity protein, BDNF, has been measured in neurally derived exosomes obtained from cord blood in babies at risk for iron deficiency and found to be suppressed(Reference Marell, Blohowiak and Evans49). This approach holds promise to link bioindicators derived from the brain and functional performance such as recognition memory capacity, which is assessable even in newborn infants.
While the utilisation of these bioindicators may never extend beyond the research realm because of cost and scalability considerations, they may be able to be linked back to the standard biomarkers of nutrient status. In the case of iron deficiency, these bioindicators change before anaemia is present. The goal would be to tie them temporally to pre-anaemic iron markers such as reticulocyte Hb per cent, serum ferritin or percentage of total iron-binding capacity saturation and utilise those markers as the scalable indices of brain iron status.
Summary
The field of nutritional neuroscience is rapidly developing with a goal of understanding how nutrients affect brain development. Nutrient effects on the brain depend on timing, dose and duration of the nutrient perturbation(Reference Kretchmer, Beard and Carlson50). The timing includes consideration of the nutrients required for critical periods of regional brain development. Certain nutrients have a high impact on early brain development and the effects of nutrient perturbations can last long after the nutrient status has normalised. The long-term effects are the true cost to society. Nutritional solutions beyond repleting deficits may be possible by leveraging emerging nutrient–brain biology. Ultimately, to judge the impact of nutrient perturbations or interventions it will be necessary to assess specific effects on brain function in human subjects.
Acknowledgements
The author would like to acknowledge Phu Tran, Sarah Cusick, Raghu Rao and Thomas Bastian for their outstanding scientific work that supported the concepts in the paper and to Daniel J Raiten for his guidance about the importance of bioindicators of nutrition.
Financial Support
The author has financial support in the form of research grants from The National Institutes of Health (USA) through the following mechanisms: R01-AA024123 (PI: Wozniak), R01-HD094809 (PI: Georgieff), R01-HD092391 (PI: Cusick), R01-NS099178 (PI: Tran).
Conflict of Interest
None.
Authorship
The author had sole responsibility for all aspects of preparation of the paper.



 Open access
Open access