


INTRODUCTION
Mosquitoes are one of the world's most recognizable insects, commonly found throughout tropical and temperate zones. Highly adaptive, these insects can also exist at high elevation and have been discovered well into the Arctic circle, although the Antarctic is currently free of their presence. The mosquitoes’ family of Culicidae sits within the order Diptera (two-winged flies) which is itself divided into two sub-families – Anophelinae and Culicinae – comprising a total of 112 genera. Their species’ biodiversity is extensive, mostly geographically structured and constantly adapting to environments to maximize reproductive output. As of February 2018, the diversity count numbered 3554 recognized species (Harbach, Reference Harbach2018).
MOSQUITO SPECIES AS A TAXONOMIC UNIT
Because mosquitoes transmit pathogens that cause disease, our studies of them are motivated not only by the desire to complete the taxonomic inventory of their biodiversity but also by their importance in the human public health arena. This emphasis has seen mosquitoes, like many species, often distinguished through the prism of the modern synthesis of evolutionary biology in which allopatric reproductive isolation leads to the emergence of distinct species (Mayr, Reference Mayr1942). These potentially interbreeding units are regarded as a fundamental category of biological organization: by definition, their mating leads to the production of fertile offspring (de Queiroz, Reference de Queiroz2005). And while reproductive isolation – whether through pre- or post-mating barriers, variance or ecological selection – can draw populations apart, if this isolation is maintained for sufficient time, post-mating barriers may develop that may include changes in chromosome organization and synteny (the localization of genes on a chromosome). This reinforces genetic isolation and completes the journey of an organism to becoming a distinct species (Ayala and Coluzzi, Reference Ayala and Coluzzi2005; Hoffmann and Rieseberg, Reference Hoffmann and Rieseberg2008).
But this journey towards to what we term ‘species’, and their genetic discontinuity, is rarely clean-cut: there can be fascinating genetic exchanges between closely related so-called ‘species’ (Mallet et al. Reference Mallet, Besansky and Hahn2016). Additionally mosquitoes often exist rather as closely related species complexes that, when studied intimately – as with the Anopheles gambiae complex in Africa – can be observed to be diverging at some parts of the genome under low recombination while other parts of the genome undergo gene flow (Besansky et al. Reference Besansky, Krzywinski, Lehmann, Simard, Kern, Mukabayire, Fontenille, Toure and Sagnon2003; Reidenbach et al. Reference Reidenbach, Neafsey, Costantini, Sagnon, Simard, Ragland, Egan, Feder, Muskavitch and Besansky2012). Do we split these into separate species or group them into one? The answer, in terms of medical entomology and vector biology, is more likely to revolve around the taxa's phenotype and its ability to transmit pathogens, adapt to control methods and exchange advantageous genes. In this way, population genetics and population genomics are revealing mosquito species’ boundaries as semipermeable, with gene exchange reflecting the genome region, blurring what are and are not fully reproductively isolated species (Crawford et al. Reference Crawford, Riehle, Guelbeogo, Gneme, Sagnon, Vernick, Nielsen and Lazzaro2015). In particular, strong selection for advantageous alleles such as insecticide resistance can result in adaptive introgressions that can breach what we understand as species’ boundaries (Norris et al. Reference Norris, Main, Lee, Collier, Fofana, Cornel and Lanzaro2015).
MOSQUITO SPECIES IDENTIFICATION
Most described species of mosquitoes have been recognized through traditional morpho-taxonomy using differential morphology as Linnaeus originally intended. Detailed morphological keys and skilled entomological technicians are often required to key out species collected in adult mosquito traps or as larvae in their aquatic environment, and regional knowledge is incredibly important to this work.
The simplest way to identify an adult mosquito is by morphology, which makes it unfortunate that mosquito scales can be easily rubbed off or are often damaged when they are collected in adult mosquito traps or not stored carefully. Perhaps even less convenient for morphological identification is the fact that mosquitoes often exist in groups of closely related sibling species in what is called a species ‘complex’ (all of which are isomorphic) or within a larger related species ‘group’ (which contains individuals with overlapping morphology that can often include a complex). The cross-mating studies that would confirm species status by observing post-mating barriers will void any natural premating barriers and are labour intensive as they usually require establishment of a colony, while the use of chromosome banding patterns to distinguish closely related cryptic Anopheles species, as pioneered the 1980s by Mario Coluzzi (Coluzzi et al. Reference Coluzzi, Sabatini, della Torre, Di Deco and Petrarca2002), is still used today (Coetzee et al. Reference Coetzee, Hunt, Wilkerson, Della Torre, Coulibaly and Besansky2013). Again, this method works best for mosquitoes that have giant polytene chromosomes – and this renders its utility outside of Anopheles limited.
Molecular genetic studies are often undertaken on mosquitoes that transmit human pathogens and these frequently discover that sibling species groups can be common within morphological taxa that can also show differences in biology (i.e. human feeding, time of feeding) and ecology (i.e. oviposition site selection, geographic distribution). These variations can result in different pathogen transmission potentials. For example, our work on the Anopleles punctulatus group of Southwestern Pacific malaria vectors over the last 20 years has used molecular genetics and the development of DNA-based species diagnostic tools to identify 13 sibling species from its three described morphological species – only five of which seem to be primary malaria vectors. [See our earlier review (Beebe et al. Reference Beebe, Russell, Burkot and Cooper2015) for a sense of this journey from the first identifications of the species as reproductively isolated, through the development of the molecular diagnostics, and the subsequent surveys that provided new insights into these malaria vectors]. The various ramifications of this in terms of vector control and the allocation of any available public health resources both underscore the crucial urgency of this type of work.
DNA BARCODES FOR MOSQUITOES
Over the past few decades, mosquito taxonomists have themselves become something of an endangered species, although whether this has led to or necessitated the shift to identifying mosquitoes using DNA sequences – more recently labelled barcodes – is a moot point. The utility of a single DNA sequence and the recent growth and utility of a mitochondrial DNA (mtDNA) cytochrome oxidase I gene (COI or COX1) sequence that can be compared universally is a reasonable starting point for categorizing mosquito biodiversity. While this review focuses mostly on the mtDNA COI, other DNA sequence targets for mosquitoes have been exploited including other mtDNA targets (ND4), nuclear gene targets (the white gene) and ribosomal DNA targets [internal transcribed spacers and ribosomal DNA (rDNA) genes]. Supplementary Table 1 provides a list of these targets and associated studies, and rDNA markers are also discussed briefly below in ‘Other Mosquito Barcodes’.
The application of the mtDNA COI barcode approach has consistently grown since its original suggestion in the early 2000s (Hebert et al. Reference Hebert, Cywinska, Ball and DeWaard2003), and the utility of using a single sequence such as the COI continues to fulfil the prerequisite for ‘Barcode of Life Data’ systems (BOLD). These are sequences that can be easily amplified with a simple protocol; their sequence region is flanked by a conserved region in which reliable primers anneal: and in this way, the organism can be capably identified at a species level. For metazoans, the COI is used; for fungi, it is the ribosomal DNA (rDNA) internal transcribed spacers (ITS); and plants utilize a multi-locus barcode (see http://www.boldsystems.org/ for more detail).
The fact that the maternally inherited COI had already enjoyed decades of use by population geneticists and molecular systematists as an evolutionary barometer contributes to its selection for this work as it appears to be an optimal tool for inferring evolutionary and demographic history as well as molecular taxonomy (Avise et al. Reference Avise, Arnold, Ball, Bermingham, Lamb, Neigel, Reeb and Saunders1987). Some of the advantages of the mtDNA COI include its universality (it is carried by all eukaryotic organisms); its relatively high copy number in the cell (which is good for PCR); and its comparatively higher substitution rate over nuclear genes (which allows for good levels of discrimination between species). It also possesses a maternal inheritance with no (or very rare) recombination in mosquitoes, and this provides a single evolutionary history. Thus, mosquitoes have become good candidates for the Barcode for Life initiative which uses a single DNA sequence to describe biodiversity by identifying species (Ratnasingham and Hebert, Reference Ratnasingham and Hebert2007).
THE MITOCHONDRIA
The mitochondria are organelles found in all eukaryotic organisms. They are likely to be a remnant ancestral bacterial endosymbiont that encodes its own independent genome of 13 coding genes – no introns are present because of its prokaryote origin, and it represents a very small fraction of the organism's actual genome size. Multiple identical copies are often present in each cell and these function as a chemical power plant for the cell generating adenosine triphosphate. For more detail on the natural history of the mitochondria – particularly with regard to its use as an evolutionary marker – see the elegant review by Ballard and Whitlock (Reference Ballard and Whitlock2004).
Multiple clonal copies within each cell make the PCR amplification of the mtDNA easier than parts of the nuclear DNA where the two paternal copies also need to be separated before sequencing. This can be achieved by either cloning or through an algorithm-driven reconstruction of paternal nuclear haplotypes post-sequencing using software like PHASE (Rozas et al. Reference Rozas, Sanchez-DelBarrio, Messeguer and Rozas2003). Separating paternal sequences with an algorithm carries with it its own issues, and these can require subsequent validation through cloning and sequencing (Garrick et al. Reference Garrick, Sunnucks and Dyer2010).
Being mostly the same clonal copy, the mtDNA is noted for having a much higher mutation rate than nuclear coding genes and this facilitates their utility as a relatively quickly evolving DNA marker (Brown et al. Reference Brown, George and Wilson1979). However, it is becoming more evident that some mosquitoes contain copies of non-functional nuclear pseudogene sequences of mitochondrial origin – or ‘numts’ (Richly and Leister, Reference Richly and Leister2004). These numts may initially appear as heteroplasmy (presenting more than one type of mitochondrial genome or sequence in an individual) and the overlapping chromatogram peaks they generate through Sanger sequencing can make subsequent analyses problematic as multiple sequences are being read as one. Nonetheless, despite increasing reports of multiple variant copies in mitochondria, which often come from the massively parallel sequencing of other organisms such as humans (Just et al. Reference Just, Irwin and Parson2015), the proportion of mosquito species with numts appears to be small overall. At present, it includes Aedes aegypti (Black and Bernhardt, Reference Black and Bernhardt2009; Hlaing et al. Reference Hlaing, Tun-Lin, Somboon, Socheat, Setha, Min, Chang and Walton2009) and the Culex pipiens group members (Behura et al. Reference Behura, Lobo, Haas, deBruyn, Lovin, Shumway, Puiu, Romero-Severson, Nene and Severson2011). Traditionally, if heteroplasmy is suspected within a species, the PCR product can be cloned and a number of clones sequenced to identify the presence of mtDNA copies. Next-generation sequencing (NGS) will also reveal heteroplasmy as it can manifest in the presence of pileup files as rare mtDNA sequences.
THE MTDNA COI BARCODE
DNA barcoding has enjoyed rapid growth as a large-scale initiative for investigating biodiversity and an audience of followers is keen to exploit its simplicity. This is despite persistent warnings from systematists who see its conceptual foundation as problematic because it stems from exclusive reliance on mitochondria (Goldstein and DeSalle, Reference Goldstein and DeSalle2011) and because of issues with inherited symbionts manipulating the maternal line (Hurst and Jiggins, Reference Hurst and Jiggins2005). The phylogenetic method normally used for these analyses is the relatively simple Neighbour-Joining (NJ) method with the Kimura 2-parameter (K2P) model initially suggested by Herbert in 2003 (Hebert et al. Reference Hebert, Cywinska, Ball and DeWaard2003). This tends to be preferred because this simpler model permits faster analyses with large datasets. Thus, the K2P model is prevalent throughout the literature, and while it assumes that transitions and transversions occur at different rates, frequencies of nucleotides are regarded as the same and an equal substitution process applies to all three codon positions (Kimura, Reference Kimura1980). The mosquito's mitochondrial genome shows the strong AT-bias and it is only really the third nucleotide within the codon (and sometimes second) that is most free to change without affecting the phenotype. With improved and more subtle evolutionary models available (Zinger and Philippe, Reference Zinger and Philippe2016), one would hope that the contemporary increased computational ability now provided by most personal computers and online servers may lead to more sophisticated evolutionary models of nucleotide evolution, given that the limitations of the K2P model may well lead to underestimations of species’ richness (Barley and Thomson, Reference Barley and Thomson2016; Zinger and Philippe, Reference Zinger and Philippe2016).
In mtDNA barcoding, there is a phenomenon known as the ‘barcoding gap’: this is the separation or distance between the mean intraspecific sequence variability and the interspecific variability for congeneric COI sequences (Meyer and Paulay, Reference Meyer and Paulay2005). If a gap exists, you can determine a cut-off value for the data identification as there would be no overlap between the interspecific and intraspecific distances. Thus the process of identifying a field-collected specimen to its species’ level is relatively straightforward if it displays minimal intraspecific variability and large interspecific variability although determining this gap requires a substantial sampling design. Some systematists favour barcode-species identifications based on only the smallest interspecific distance as the mean interspecific distances are artificially inflated – see work by Meier et al. for more detail on the differentiation methodologies of species (Meier et al. Reference Meier, Zhang and Ali2008).
BROAD-SCALE COI BARCODING
Before the mtDNA COI was employed as a standalone entity, it was often used alongside nuclear DNA sequence regions in studies of mosquito biodiversity, and this approach allowed sibling species to be teased apart in order to study their biology, behaviour and pathogen transmission potential (see the supplementary Table 1 for information on how the COI has been co-assessed alongside other markers in mosquito studies). Although the COI was used earlier as a population genetics tool in this way, the barcode concept itself was substantially forged in the 21st century (Hebert et al. Reference Hebert, Cywinska, Ball and DeWaard2003).
The first mosquito study to employ a dedicated COI barcoding approach came from researchers involved in DNA barcoding (Cywinska et al. Reference Cywinska, Hunter and Hebert2006). Using mosquito collections from Canada that had initially been identified to species by morphology, they combined this information with additional mosquito COI sequences pulled from Genbank (Cywinska et al. Reference Cywinska, Hunter and Hebert2006). Again, a relatively simple model of evolution promoting computational speed was used – an NJ analysis with K2P (Kimura, Reference Kimura1980). Outcomes from this study were reasonably compelling for species that were distantly related within a genus and barcode congruence (evidence of the same sequences) was found between some closely related species that were still morphologically distinct. Interesting data from that first study suggests that 98% of mosquito species were <2% divergent. The small divergence recorded may reflect the limited sampling of individuals within species, although one Aedes species encountered in this work was found to be 3.6–3.9% divergent. Surprisingly only one pseudogene generated detectable numts, and this was encouraging because it again suggested that numts may not be common in mosquitoes.
Indeed, morphological and molecular comparisons of the COI sequence do appear congruent in studies across genus levels, as attested by morphological species’ studies on mosquito diversity from Argentina (Laurito et al. Reference Laurito, de Oliveira, Almiron and Sallum2013), Australia (Batovska et al. Reference Batovska, Blacket, Brown and Lynch2016), China (Wang et al. Reference Wang, Li, Guo, Xing, Dong, Wang, Zhang, Liu, Zheng, Zhang, Zhu, Wu and Zhao2012), India (Kumar et al. Reference Kumar, Rajavel, Natarajan and Jambulingam2007), Singapore (Chan et al. Reference Chan, Chiang, Hapuarachchi, Tan, Pang, Lee, Lee, Ng and Lam-Phua2014), Italy (Talbalaghi and Shaikevich, Reference Talbalaghi and Shaikevich2011), Iran (Azari-Hamidian et al. Reference Azari-Hamidian, Yaghoobi-Ershadi, Javadian, Abai, Mobedi, Linton and Harbach2009) and Pakistan (Ashfaq et al. Reference Ashfaq, Hebert, Mirza, Khan, Zafar and Mirza2014). This latter study from Pakistan reveals intraspecific divergences at a maximum of 2.4% from over 1600 individuals from 24 taxa – a result in line with the original Canadian work. In addition to this, the COI barcode appears to complement taxa described by morphology within the generic and subgeneric levels where morphology still has an important utility (Torres-Gutierrez et al. Reference Torres-Gutierrez, Bergo, Emerson, de Oliveira, Greni and Sallum2016). Most of these studies were performed on endemic species (with the necessary inclusion of some ubiquitous exotics), and the utility of the COI barcode as a correlate with morphology is perhaps most valuable at this relatively broadscale level: it complements traditional morphological taxonomy in cataloguing mosquitoes from regional landscapes. Unfortunately, many of these studies employ only a small intraspecific sample size and this may deliver a biased picture of the intraspecific variation within these taxonomic units by conveying inaccurate intraspecific divergences. Because of this, sampling across the full range of the species is advised. Even museum specimens can now be regarded as biobanks as new NGS technologies can obtain adequate molecular data from old specimens and overcome issues with DNA damage in them as well (Yeates et al. Reference Yeates, Zwick and Mikheyev2016).
FINE-SCALE COI BARCODING
Moving on from these comparisons with taxonomy by morphology where the COI barcode and morphology appear to correlate relatively well, we travel into the darkness of closely related sibling and cryptic species groups and complexes, where species morphology is either polymorphic for diagnostic characters or isomorphic, with multiple species hidden under the same morphology. In this area, a barcode can indeed shed important light on divergent lineages or hypothetical species that may represent reproductively isolated taxa. For example, studies on Culex species in Australasia have identified novel divergent lineages, one of which correlates to the southern limit of Japanese encephalitis activity in the region (Hemmerter et al. Reference Hemmerter, Slapeta, van den Hurk, Cooper, Whelan, Russell, Johansen and Beebe2007). When this study was followed up with a nuclear sequence from the acetylcholine esterase 2 (ace2), the nuclear marker supported the discovery of species’ level reproductive isolation (Hemmerter et al. Reference Hemmerter, Slapeta and Beebe2009).
However not all investigations deliver such clear-cut results: a case study from Argentina and Brazil that sought to resolve several Culex taxa using the COI only managed to resolve 69% of species with the remaining unresolved individuals registering as ambiguous (10%), misidentified (18%), or unidentified (3%) (Laurito et al. Reference Laurito, de Oliveira, Almiron and Sallum2013). Recently diverged Culex mosquitoes, especially those in the Cx. pipiens group, can show insufficient variation at either the COI or the rDNA ITS2 for classification (Crabtree et al. Reference Crabtree, Savage and Miller1995; Danabalan et al. Reference Danabalan, Ponsonby and Linton2012; Batovska et al. Reference Batovska, Cogan, Lynch and Blacket2017) and they are notorious for showing hybrids where species’ distributions overlap (Farajollahi et al. Reference Farajollahi, Fonseca, Kramer and Marm Kilpatrick2011; Tahir et al. Reference Tahir, Kanwal and Mehwish2016).
But at this finer scale, and with good field sampling, the COI barcode has proved useful in recent anopheline biodiversity studies in both Africa and the Western Pacific (Lobo et al. Reference Lobo, St Laurent, Sikaala, Hamainza, Chanda, Chinula, Krishnankutty, Mueller, Deason, Hoang, Boldt, Thumloup, Stevenson, Seyoum and Collins2015; Laurent et al. Reference Laurent, Cooke, Krishnankutty, Asih, Mueller, Kahindi, Ayoma, Oriango, Thumloup, Drakeley, Cox, Collins, Lobo and Stevenson2016). Given that it is maternally inherited through the female egg – and so is excluded from the direct influence of sex – the mtDNA cannot reveal reproductive isolation like the nuclear DNA. It can, however, reveal the presence of divergent lineages reflecting the kind of long-standing isolation that permits mtDNA lineages to fully differentiate into separate genetic clades (lineage sorting). On the other hand, problems may manifest when mtDNA markers are used to discriminate recently diverges species. We see this phenomenon in well-studied groups of cryptic mosquito species such as the African An. gambiae complex where lineages have not fully sorted into divergent clades or where mtDNA introgression may still be shuffling mitochondria between what we have previously called distinct species (Thelwell et al. Reference Thelwell, Huisman, Harbach and Butlin2000; Donnelly et al. Reference Donnelly, Pinto, Girod, Besansky and Lehmann2004). Indeed the An. gambiae complex gives us one of the best illustrations of the complexity of determining reproductive isolation in recently diverged cryptic species as it permits us to peer into the fascinating evolutionary dynamics occurring in diverging populations (Weetman et al. Reference Weetman, Steen, Rippon, Mawejje, Donnelly and Wilding2014; Mallet et al. Reference Mallet, Besansky and Hahn2016). The An. gambiae complex is likely the rule rather than the exception of what can be occurring in recently diverged populations.
Despite the mitochondrial genome displaying in general a higher mutation rate and smaller effective population size (one copy of the genome) than the nuclear genome (which delivers four copies of autosomal nuclear DNA to the next generation), the mtDNA should in theory fix alleles much faster than nuclear DNA (Ballard and Whitlock, Reference Ballard and Whitlock2004). Nonetheless, incomplete lineage sorting of COI sequences between recently diverged species can result in species being overlooked because shared COI sequences still exist in both populations, as either ancestral haplotypes or through interspecies introgression events between related species (Donnelly et al. Reference Donnelly, Pinto, Girod, Besansky and Lehmann2004; Bennett et al. Reference Bennett, Linton, Shija, Kaddumukasa, Djouaka, Misinzo, Lutwama, Huang, Mitchell, Richards, Tossou and Walton2015; Surendran et al. Reference Surendran, Truelove, Sarma, Jude, Ramasamy, Gajapathy, Peiris, Karunaratne and Walton2015). Figure 1 provides a simple graphical example of where barcode sharing occurs in the study of closely related or recently diverged species.
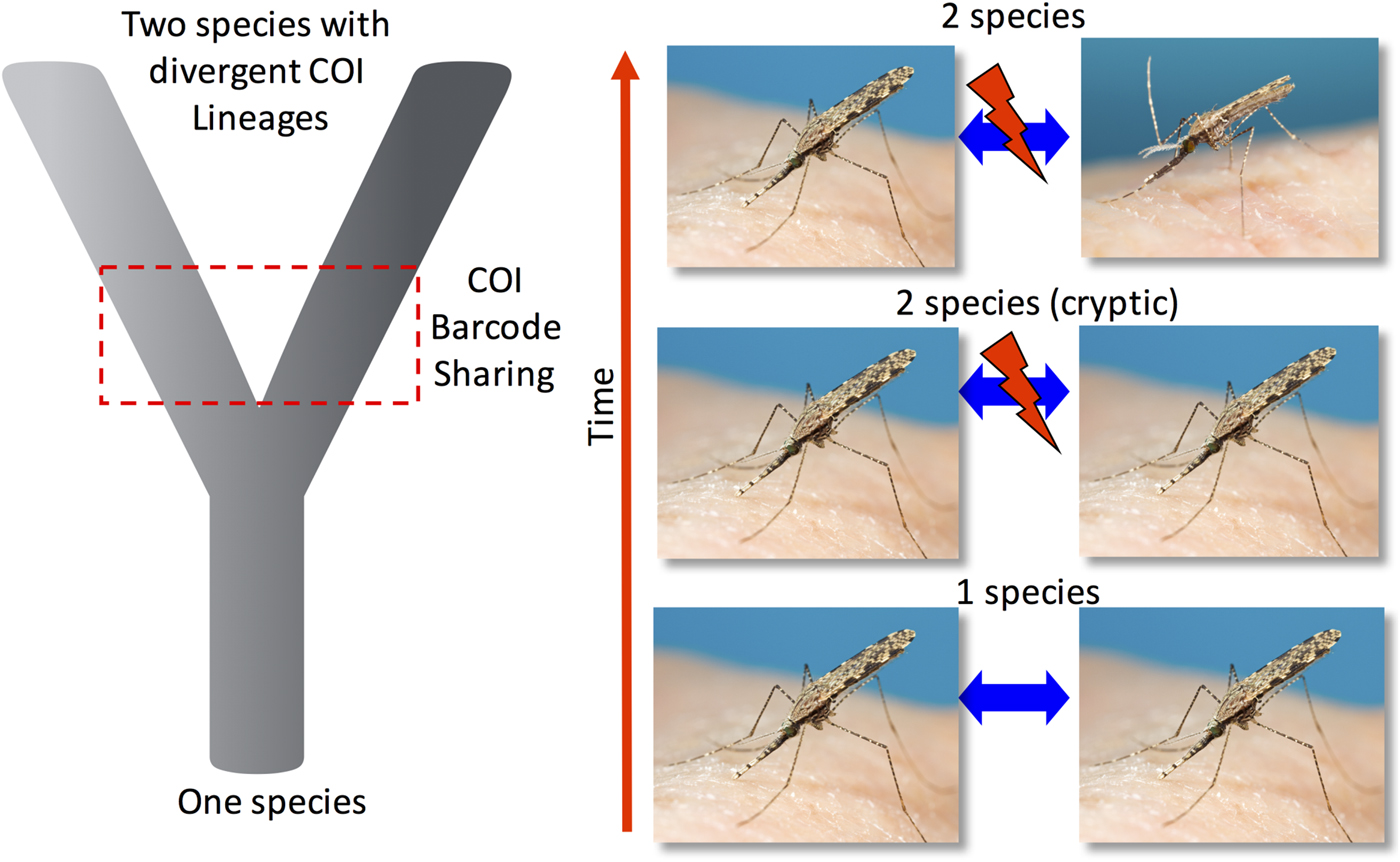
Fig. 1. A simplified representation of the progression of one species diverging into two reproductively isolated species. Often there is a period of time where the two recently discrete species are regarded as morphologically cryptic and cannot be distinguished by external morphology. During early stage of isolation shared mtDNA COI sequences can still be present and have not been fully sorted into discrete mtDNA COI lineages despite being reproductively incompatible. Additionally, mitochondrial genomes can be introgressed between closely related species through hybridization again resulting in mtDNA barcode sharing.
The mitochondria organelle can show metabolic differences that affect the phenotype, and experiments in Drosophila suggest its electron transport system is not impaired by introgression events (Pichaud et al. Reference Pichaud, Ballard, Tanguay and Blier2012), and so may facilitate the uptake of more fit mitochondria through such introgression events. If the mitochondria provide a selective advantage to the organism, the introgressed mitochondria can sweep through a population or species obscuring the evolutionary signal by placing individuals in an incorrect taxonomic clade. For example, an evolutionary study of ours on two sympatric Anopheles species from the Southwest Pacific identified what appears to be a mitochondrial sweep from an introgression event between two related species (Ambrose et al. Reference Ambrose, Riginos, Cooper, Leow, Ong and Beebe2012). Figure 2 depicts this scenario where a genetically and geographically restricted population of the coastal restricted species Anopheles farauti appears paraphyletic for the COI in the phylogeny, and this introgressed population sits well within its sister species Anopheles hinesorum, which also occurs inland and at elevation. Both species are reciprocally monophyletic for nuclear markers, but the An. hinesorum-like mtDNA sequences found in the An. farauti appear to have swept through the large population that spans from northeast Australia and southern New Guinea. If we had only used the COI marker on these populations, we would have mistakenly thought that An. hinesorum is saline water tolerant through some of its range, but this is not the case: only An. farauti is saline water tolerant (Sweeney et al. Reference Sweeney, Cooper and Frances1990).
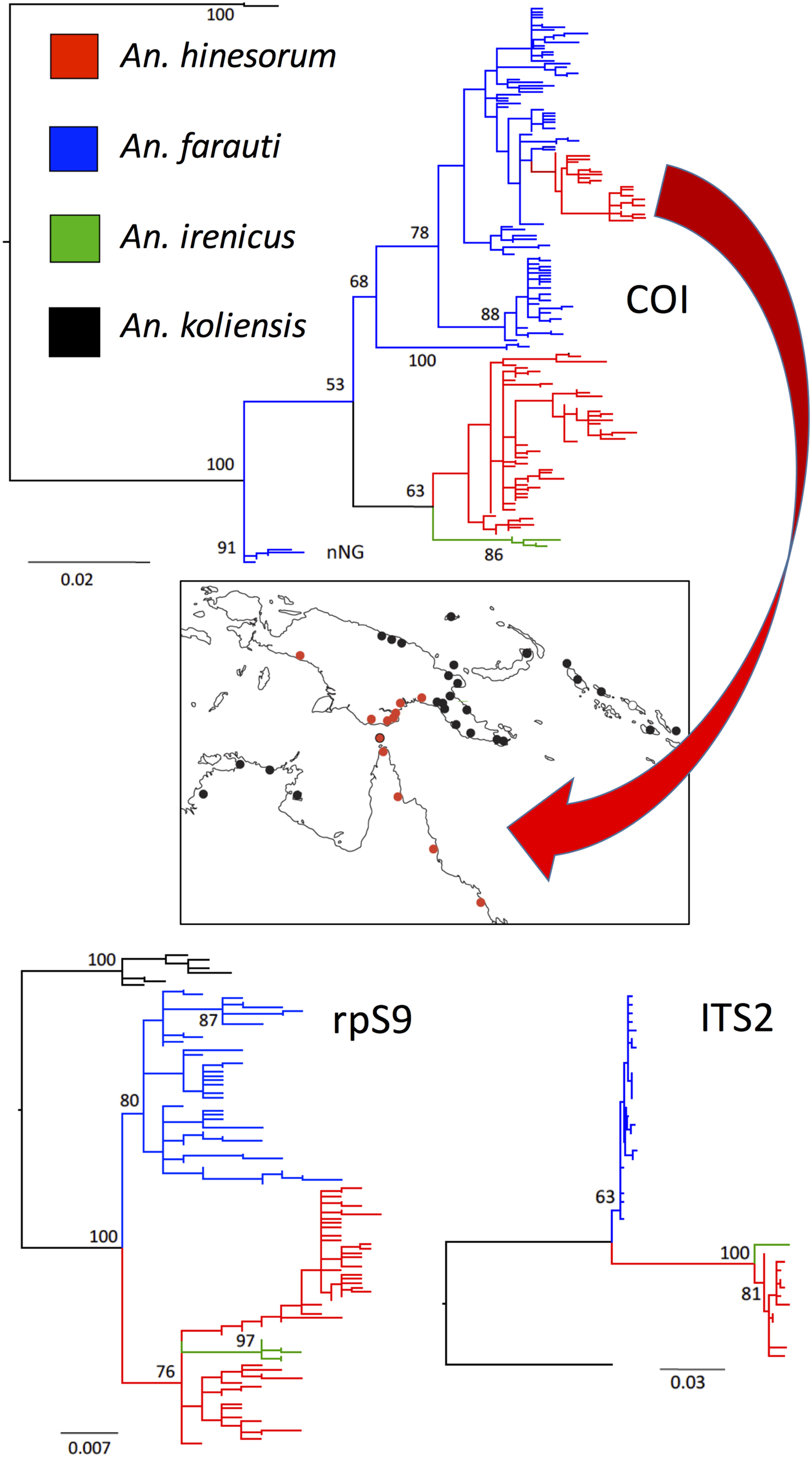
Fig. 2. Potential conflicting scenarios between the mtDNA and nuclear DNA. Cryptic species An. fatrauti (coastal), An. hinesorum (coastal and inland), An irenicus (Solomon Islands restricted) and outgroup An. koliensis (New Guinea) were sequenced for the mtDNA COI and two nuclear markers [ITS2 and ribosomal protein S9 (rpS9)]. A PhyML analysis reveals that An. farauti and An. hinesorum are reciprocally monophyletic at both nuclear markers; however, a genetic and geographic population of An. farauti also appears paraphyletic for the mtDNA COI. This An. farauti COI population emerges within An. hinesorum (red branch in blue An. hinesorum tree). The distribution of this mtDNA population is also shown on the map in red and is most likely the result of past introgression with An. hinesorum followed by a mitochondrial sweep through a genetically and geographically restricted An. farauti population in northwest Australia and southern New Guinea (figure was modified from our study of Ambrose et al. Reference Ambrose, Riginos, Cooper, Leow, Ong and Beebe2012).
INDIRECT SELECTION ON MTDNA
Arthropods often carry passenger microorganisms that exist in their cells and are passed from a female to her progeny through an egg. This phenomenon – whether its results are positive or negative – can place an indirect selection pressure on the mtDNA arising from linkage disequilibrium with the maternally inherited arthropod symbionts (Hurst and Jiggins, Reference Hurst and Jiggins2005). In mosquitoes, the effects of the maternally inherited symbiont Wolbachia on the evolution of mtDNA can be seen through the indirect selection they place on the mtDNA sequences/haplotype that will co-migrate through the maternal line. A neat example of this phenomenon was observed in an early study on Drosophila from Californian populations of D. simulans where a strain of Wolbachia swept through an uninfected population during the 1980s (at 100 km year−1) driven by the Wolbachia’s cytoplasmic incompatibility with uninfected wild types (Turelli et al. Reference Turelli, Hoffmann and Mckechnie1992). This led to the transformation of the population's mtDNA as it hitchhiked along with the symbiont. While a mechanism for Wolbachia cytoplasmic incompatibility has only recently been described (Beckmann et al. Reference Beckmann, Ronau and Hochstrasser2017; LePage et al. Reference LePage, Metcalf, Bordenstein, On, Perlmutter, Shropshire, Layton, Funkhouser-Jones, Beckmann and Bordenstein2017), the symboint's ability to obscure the mtDNA evolutionary signal through selected sweeps has been described in other Dipteran species such as fruit flies, where it reduces mtDNA diversity and drives new or rare haplotypes through populations (Whitworth et al. Reference Whitworth, Dawson, Magalon and Baudry2007; Nunes et al. Reference Nunes, Nolte and Schlotterer2008; Schuler et al. Reference Schuler, Koppler, Daxbock-Horvath, Rasool, Krumbock, Schwarz, Hoffmeister, Schlick-Steiner, Steiner, Telschow, Stauffer, Arthofer and Riegler2016). Outside of the Cx. pipiens group of mosquitoes (Rasgon et al. Reference Rasgon, Cornel and Scott2006), this phenomenon has not been well described, however artificially transformed Wolbachia infected Ae. aegypti are successfully being used to transform wild Ae. aegypti populations in a bid to reduce virus transmission (O'Neill, Reference O'Neill2016), and it will be interesting to watch both the outcomes of these artificially induced selective sweeps and the ultimate genetic changes within the transformed and untransformed Ae. aegypti populations (Yeap et al. Reference Yeap, Rasic, Endersby-Harshman, Lee, Arguni, Le Nguyen and Hoffmann2016).
OTHER MOSQUITO BARCODES: RIBOSOMAL DNA
A PubMed search of literature about the utility of a DNA barcode using the terms ‘mosquito, COI and species’ calls up more than 150 papers. But recasting this search reveals that the most common DNA marker for mosquito identification has been the rDNA ITS2 (replacing COI with ITS2 as a search term). PubMed cites 223 papers that have employed this marker either alone or alongside other, of which 193 (~90%) have been on anopheline mosquitoes. These papers include studies on the spacers’ utility as PCR-based tools for species diagnostics methods where the goal is not DNA sequencing but rather DNA genotyping for species based on specific polymorphisms that can provide allele-specific primers (Porter and Collins, Reference Porter and Collins1991), or on restriction analyses of PCR products (Beebe and Saul, Reference Beebe and Saul1995). It is important to note, too, that while the rDNA large subunit D2 and D3 regions are also used, the ITS2 appears to be the most published molecular marker for mosquitoes. The rDNA D2 and D3 subunit are part of the structural RNA gene, while the ITS2 is an intriguingly expedient spacer that separates two structural RNA genes (5.8S and 28S). Spliced out of the mature RNA, it appears to accommodate mutations more quickly than gene regions. Figure 3A gives a simplistic illustration of the rDNA gene family organization and the relative positioning of these markers.

Fig. 3. The ribosomal DNA (rDNA) spacers have been popular as a genetic marker for species-level studies. The ribosomal gene family array [panel (A), rDNA unit] sits tandemly organized head to tail in the array, often 100 s of times usually positioned at a single locus in mosquitoes. The units in the array are observed to evolve together (in concert) although sequence variant copies can manifest in the array causing problems with traditional Sanger sequencing. These variant copies in the array can be seen by a simple acrylamide electrophoresis of the PCR product as variant copies that form mispaired double-stranded DNA duplexes (heteroduplexes) retard migration through the gel-forming laddering effect – more paralogues (variants) the more laddering occurs. Direct sequencing can fail and cloning was usually required prior to sequencing.
Yet in many ways, the rDNA provides a peculiar DNA barcode target to use for species identification. It exists in the genome as a multicopied tandem gene family array, up to hundreds of copies in the metazoan genome, and it evolves through a non-Mendelian process (Dover, Reference Dover2002). This evolutionary process is still not well described in and of itself: various theories to explain this pattern of concerted evolution where gene family units evolve together have been proposed, but we still do not have a unifying theory for this gene family evolution (Dover, Reference Dover2002; Nei and Rooney, Reference Nei and Rooney2005; Eickbush and Eickbush, Reference Eickbush and Eickbush2007). But despite the current deficiencies in detailing the rDNA evolutionary process, its intimate involvement in sex and its rapid evolutionary turnover give it crucial utility as a species-level marker because of its ability to manifest early genetic discontinuities (Bower et al. Reference Bower, Dowton, Cooper and Beebe2008), and to reveal cryptic species-level diversity (Paskewitz et al. Reference Paskewitz, Wesson and Collins1993). Thus in many ways, the rDNA maintains a utility over the mtDNA COI in terms of the speed of lineage sorting of its multicopy rDNA array. As a comparable evolutionary marker for closely related species, rDNA's utility is often complementary to the COI (Alquezar et al. Reference Alquezar, Hemmerter, Cooper and Beebe2010; Ruiz-Lopez et al. Reference Ruiz-Lopez, Wilkerson, Ponsonby, Herrera, Sallum, Velez, Quinones, Flores-Mendoza, Chadee, Alarcon, Alarcon-Ormasa and Linton2013; Lobo et al. Reference Lobo, St Laurent, Sikaala, Hamainza, Chanda, Chinula, Krishnankutty, Mueller, Deason, Hoang, Boldt, Thumloup, Stevenson, Seyoum and Collins2015). However, as it is rare to find heteroplasmy in the mitochondrial marker, it is not uncommon to observe intragenomic copy variants appearing in mosquito rDNA sequencing, making direct PCR-Sanger sequencing tricky to read and often causing chromatograms to collapse (Bower et al. Reference Bower, Dowton, Cooper and Beebe2008; Alquezar et al. Reference Alquezar, Hemmerter, Cooper and Beebe2010; Batovska et al. Reference Batovska, Cogan, Lynch and Blacket2017). Because intra-individual sequence variants in rDNA copies that contain insertion/deletion indel variants can cause chromatograms to collapse, cloning prior to sequencing is often required. We have found that the decision to clone or not to clone can be assessed by running the rDNA PCR product through a native acrylamide gel: paired strands between different sequence variants can be visualized as they migrate more slowly in the gel. [See Fig. 3B for a graphic portrayal of visualizing intragenomic ITS2 variants as well as the following citations (Beebe et al. Reference Beebe, Maung, van den Hurk, Ellis and Cooper2001; Alquezar et al. Reference Alquezar, Hemmerter, Cooper and Beebe2010).]
The answer as to why the rDNA is so prevalent as a species diagnostic target for mosquitoes is probably due to its high rate of mutation and rapid DNA turnover within and between rDNA repeats (Eickbush and Eickbush, Reference Eickbush and Eickbush2007; Bower et al. Reference Bower, Cooper and Beebe2009; Alquezar et al. Reference Alquezar, Hemmerter, Cooper and Beebe2010). The rDNA gene family in Anopheles mosquitoes is positioned near the centromere on the sex chromosomes (Kumar and Rai, Reference Kumar and Rai1990). The reduced the rate of recombination in this genomic landscape may influence the rate of genetic divergence (Nachman and Churchill, Reference Nachman and Churchill1996; Stump et al. Reference Stump, Fitzpatrick, Lobo, Traore, Sagnon, Costantini, Collins and Besansky2005). In this, genetic divergence can be observed to manifest, particularly in the rDNA in the face of apparent gene flow at other parts of the genome (Slotman et al. Reference Slotman, Reimer, Theimann, Dolo, Fondjo and Lanzaro2006; Weetman et al. Reference Weetman, Wilding, Steen, Pinto and Donnelly2012). Despite much of the published literature being based on Anopheles mosquitoes, the rDNA ITS2 spacer appears to also perform well as a species level marker with Culex mosquitoes (Vesgueiro et al. Reference Vesgueiro, Demari-Silva, Malafronte, Sallum and Marrelli2011). The rDNA ITSI and ITS2 display utility as molecular diagnostics targets for other mosquito genera (Beebe et al. Reference Beebe, van den Hurk, Chapman, Frances, Williams and Cooper2002; Beebe et al. Reference Beebe, Whelan, Van den Hurk, Ritchie, Corcoran and Cooper2007; Higa et al. Reference Higa, Toma, Tsuda and Miyagi2010; Montgomery et al. Reference Montgomery, Shivas, Hall-Mendelin, Edwards, Hamilton, Jansen, McMahon, Warrilow and van den Hurk2017). The fast-evolving rDNA transcribed spacers (such as the ITS2) have thus become useful markers for revealing early genetic discontinuities in populations and for providing species-level discrimination.
One must advocate caution as DNA sequence-based identification and the high rate of rDNA spacer evolution (turnover) can lead to geographically structured populations developing distinct ITS2 sequences as found in An. farauti in the Western Pacific (Beebe et al. Reference Beebe, Cooper, Morrison and Ellis2000; Bower et al. Reference Bower, Dowton, Cooper and Beebe2008). The inability or perhaps complexity of detecting the presence of shared heterozygotes in these multigene family situations makes assessing gene flow signatures between geographically separated populations tricky. Indeed with sequence variation within seemingly identical species found in a Latin American anophelines study that used submitted GenBank ITS2 sequences revealed intraspecific variation ranging from 0.2 to 19% (Marrelli et al. Reference Marrelli, Sallum and Marinotti2006). The authors’ wisely caution that a minimum requirement be considered for additional studies include voucher specimens, sampling for intraspecific variation and the use of other molecular markers.
For more details on the utility of these and other molecular markers used to study mosquitoes, a supplementary table (supplementary Table 1) is available as a summary of PubMed searches on genetic markers for mosquito identification. This table – of over 200 mosquito studies – is not exhaustive but provides an insight into the diversity of genomic regions used for taxonomic and genetic analyses of mosquito species and the ITS2 is by far the most commonly used. Indeed, the size of the ITS regions may have some bearing on the ability of the sequence to acquire non-deleterious mutations with longer sequences generally better able to accommodate changes than shorter sequences (Alquezar et al. Reference Alquezar, Hemmerter, Cooper and Beebe2010). This effect whereby ITS length can accommodate larger amounts of variation can be observed in studies of the neighbouring ITS1 where lengths can exceed 2000 bp and show large amounts of intraindividual and intraspecific variation that may be difficult to manage (Bower et al. Reference Bower, Dowton, Cooper and Beebe2008, Reference Bower, Cooper and Beebe2009).
In regard to aligning rDNA spacer sequences, rDNA spacer sequences can contain large amounts of indels and repeat sequences and these can be tricky to align. Thus, computer-based alignments may require editing by eye (Beebe et al. Reference Beebe, Cooper, Morrison and Ellis2000; Bower et al. Reference Bower, Dowton, Cooper and Beebe2008, Reference Bower, Cooper and Beebe2009). Fortunately, there is now a ‘How to’ manual for molecular systematics to assist with ITS2 sequence alignments and to guide sequence alignments based on secondary structure (Schultz and Wolf, Reference Schultz and Wolf2009).
SEQUENCING DNA BARCODES
While Sanger sequencing now provides a relatively cheap and simple means to acquire individual sequences (~$US5.00/sequence at the time of writing), the advent of next-generation sequencing (NGS) platforms permits the parallel acquisition of DNA barcode sequences from numerous specimens simultaneously – after initial morphological classification down to species or species complex. These methods have been assessed and compared with traditional Sanger sequencing and found to be both superior and more efficient in terms of labour and cost (Shokralla et al. Reference Shokralla, Gibson, Nikbakht, Janzen, Hallwachs and Hajibabaei2014; Shokralla et al. Reference Shokralla, Porter, Gibson, Dobosz, Janzen, Hallwachs, Golding and Hajibabaei2015; Batovska et al. Reference Batovska, Cogan, Lynch and Blacket2017).
The first method employed 454 pyrosequencing on 190 Lepidoptera specimens to recover, after bioinformatics analysis, full-length DNA barcodes. Only 12.5% of a 454 sequencing run's capacity had to be utilized to provide 143 sequence reads for each specimen. When compared with Sanger sequencing of the same 190 individuals – which delivered longer individual reads for each specimen than the 454 sequencing – the 454 showed a superior ability to discriminate species number, heteroplasmic sequences and nuclear mtDNA introgressions (Shokralla et al. Reference Shokralla, Gibson, Nikbakht, Janzen, Hallwachs and Hajibabaei2014). The second method used a double dual-indexing approach on an Illumina MiSeq to identify 1010 specimens from 11 orders of arthropods collected from a single Malaise trap sample from Area de Conservación Guanacaste in northwestern Costa Rica (Shokralla et al. Reference Shokralla, Porter, Gibson, Dobosz, Janzen, Hallwachs, Golding and Hajibabaei2015). Again, this alternative method proved better than Sanger sequencing of COI barcodes with the authors able to cite a 27% reduction in total time required, a 78% reduction in hands-on time, and a 79% reduction in laboratory costs. A similar Illumina MiSeq-based method has also been used for sequencing the ITS2 barcode of 26 species of mosquitoes collected from Australia which were compared to Sanger sequencing of the same samples (Batovska et al. Reference Batovska, Cogan, Lynch and Blacket2017). The authors of this study also found superior resolution compared to the Sanger sequencing on the same individuals and could avoid the common difficulty of Sanger sequence chromatograms collapsing when individuals contain multiple ITS2 sequence variants with insertion/deletion indels.
The three methods described above all utilized PCR amplification of the barcodes regions prior to sequencing. Perhaps as NGS costs reduce, low-level genome skims using whole genomic DNA from individual specimens will prove to be the future for barcoding (Crampton-Platt et al. Reference Crampton-Platt, Yu, Zhou and Vogler2016), permitting the full reconstruction of high-copy DNA such as the mtDNA and rDNA.
CONCLUDING REMARKS
This review has focused mostly on the use of mtDNA COI DNA barcode for species identification of mosquitoes given that its utility for the study of mosquitoes is growing rapidly. For closely related species such as cryptic species groups and complexes, investigators should proceed with caution given problems with incomplete lineage sorting and introgression events where the same COI sequence may still appear in different species or may introgress across what we take to be species’ boundaries. In all cases, it is advisable to run a nuclear marker alongside the mtDNA, and the rDNA – in particular the ITS2 – can provide a useful counterpoint to the COI (Alquezar et al. Reference Alquezar, Hemmerter, Cooper and Beebe2010; Ajamma et al. Reference Ajamma, Villinger, Omondi, Salifu, Onchuru, Njoroge, Muigai and Masiga2016).
We are only at the beginning of this journey in linking mosquito species using initial morphological taxonomy with molecular characters. With there currently being ~3500 described species (Harbach, Reference Harbach2017), there is a paucity of molecular data available on these species and one would invisage this species number esculating as the many cryptic and undescribed species are assembled. It is useful to distinguish DNA barcoding from DNA-based taxonomy – both of which were proposed to support inefficiencies and difficulties using traditional morphology-based taxonomy and to permit non-taxonomists to develop species’ identification methods and tools. We often now see a process of taxonomic understanding that combines morphological and molecular data that come together as an ‘integrative taxonomy’ (Teletchea, Reference Teletchea2010). It seems important to reiterate that the Linnaean system of nomenclature should always be fundamental to describing the hierarchy of biodiversity down to species, and following the journey from primary literature through taxa classification by description and revision is paramount to any working understanding of species. This should also continue as advances in DNA sequencing generate hypotheses for the discovery and delineation of new species. More useful information on describing biodiversity can be found in reviews by Goldstein and DeSalle, and Kress, Garcia-Robledo et al. (Goldstein and DeSalle, Reference Goldstein and DeSalle2011; Yeates et al. Reference Yeates, Seago, Nelson, Cameron, Joseph and Trueman2011; Kress et al. Reference Kress, Garcia-Robledo, Uriarte and Erickson2015). A general warning is probably useful here as the available databases are likely littered with poorly identified species and incorrect sequences so investigators should beware of spurious hits from incorrectly identified species sequences. Finally, the move to next generation sequencing for mosquito barcoding is exciting as it would allow researchers to run multiple target barcodes for less cost and effort (Batovska et al. Reference Batovska, Cogan, Lynch and Blacket2017). The sharing of bioinformatics pipelines would be strongly encouraged.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182018000343.
ACKNOWLEDGEMENTS
The author would like to thank David Yeates for comments on the manuscript and Steve Doggett for the mosquito photos used in Fig. 1.
FINANCIAL SUPPORT
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.






 Open access
Open access