


INTRODUCTION
Many factors have been proposed to explain parasite diversity (Poulin, Reference Poulin1997). For example, previous studies have shown that species richness increases towards the equator for some groups of parasitic organisms (Rohde and Heap, Reference Rohde and Heap1998; Guernier et al. Reference Guernier, Hochberg and Guégan2004; Nunn et al. Reference Nunn, Altizer, Sechrest and Cunningham2005). However, a recent meta-analysis of 62 studies involving animal, plant and fungal hosts showed that there was no strong evidence for an effect of latitude on parasite species richness (Kamiya et al. Reference Kamiya, O'Dwyer, Nakagawa and Poulin2014). Parasite diversity might be determined by characteristics of hosts rather than those of the environment. For example, host body size, population density and geographic range have all been suggested as universal predictors of variation in parasite species richness (Kamiya et al. Reference Kamiya, O'Dwyer, Nakagawa and Poulin2014). Nevertheless, the meta-analysis of Kamiya et al. (Reference Kamiya, O'Dwyer, Nakagawa and Poulin2014) did not include vector-transmitted parasites. Host density can be especially important for parasites, which depend on hematophagous insects for reproduction because the concentration of hosts in a given area potentially affects the prevalence and transmission of vector-borne pathogens by influencing encounter rates between vectors and susceptible hosts (Nunn and Heymann, Reference Nunn and Heymann2005).
Avian malaria is a worldwide, vector-transmitted disease caused by haemosporidian parasites in the genus Plasmodium (Valkiūnas, Reference Valkiūnas2005). These parasites reproduce sexually in female mosquito vectors from the genera Culex, Aedes, Culiseta, Anopheles, Mansonia, Aedeomyia and Coquillettidia (Diptera: Culicidae) (Valkiūnas, Reference Valkiūnas2005; Njabo et al. Reference Njabo, Cornel, Sehgal, Loiseau, Buermann, Harrigan, Pollinger, Valkiūnas and Smith2009; Santiago-Alarcon et al. Reference Santiago-Alarcon, Palinauskas and Schaefer2012). Environmental factors, especially temperature, can play a role in the distribution, prevalence and transmission of these parasites (Gonzalez-Quevedo et al. Reference Gonzalez-Quevedo, Davies and Richardson2014; Oakgrove et al. Reference Oakgrove, Harrigan, Loiseau, Guers, Seppi and Sehgal2014). Temperature constrains not only parasite sporogonic development (LaPointe et al. Reference LaPointe, Goff and Atkinson2010), but also influences the activity and development of the mosquito vectors, which are important determinants of the prevalence and transmission of avian malaria. However, temperature, among other environmental variables, did not explain avian malaria prevalence in avian species across several sites in South Africa (Okanga et al. Reference Okanga, Cumming and Hockey2013). As Okanga et al. (Reference Okanga, Cumming and Hockey2013) pointed out, this suggests that the prevalence of avian malaria parasites may also be determined by factors related to their avian hosts and mosquito vectors (see also Ellis et al. Reference Ellis, Collins, Medeiros, Sari, Coffey, Dickerson, Lugarini, Stratford, Henry, Merrill, Matthews, Hanson, Roberts, Joyce, Kunkel and Ricklefs2015).
Several studies have investigated the effect of avian ecological traits on the probability of infection by Plasmodium and other related haemosporidians (Ricklefs et al. Reference Ricklefs, Swanson, Fallon, Martinez-Abrain, Scheuerlein, Gray and Latta2005; Fecchio et al. Reference Fecchio, Lima, Silveira, Braga and Marini2011, Reference Fecchio, Lima, Svensson-Coelho, Marini and Ricklefs2013; Svensson-Coelho et al. Reference Svensson-Coelho, Blake, Loiselle, Penrose, Parker and Ricklefs2013; González et al. Reference González, Matta, Ellis, Miller, Ricklefs and Gutiérrez2014; Lutz et al. Reference Lutz, Hochachka, Engel, Bell, Tkach, Bates, Hackett and Weckstein2015; Matthews et al. Reference Matthews, Ellis, Hanson, Roberts, Ricklefs and Collins2016). In these studies, variation in haemosporidian prevalence is thought to be a result of the host's capability to resist and control infection or the result of differential exposure to parasites. However, most studies have not considered the role of vectors in explaining these patterns (but see Medeiros et al. Reference Medeiros, Ricklefs, Brawn and Hamer2015).
At a finer spatial scale, infection risk for Plasmodium in blue tit populations increased with increasing proximity to a large water source, possibly as result of increased vector abundance (Wood et al. Reference Wood, Cosgrove, Wilkin, Knowles, Day and Sheldon2007). Haemosporidian prevalence was also higher in the wettest of two western Amazonian 100 ha forest plots that were otherwise similar with respect to forest type, altitude, human disturbance and flooding (Svensson-Coelho et al. Reference Svensson-Coelho, Blake, Loiselle, Penrose, Parker and Ricklefs2013). These studies corroborate the idea that prevalence of blood parasites is higher at sites where vectors are more abundant.
Besides vector abundance, avian host density could also play a role in determining the prevalence of avian malaria parasites. For example, host population density can influence the spread and distribution of parasites by increasing the probability that the vectors and thus the parasites can come into contact with hosts (Anderson and May, Reference Anderson and May1978; Ellis et al. Reference Ellis, Medeiros, Collins, Sari, Coffey, Dickerson, Lugarini, Stratford, Henry, Merrill, Matthews, Hanson, Roberts, Joyce, Kunkel and Ricklefs2017). Furthermore, Drovetski et al. (Reference Drovetski, Aghayan, Mata, Lopes, Mode, Harvey and Voelker2014) found that haemosporidian lineages infected abundant bird species more frequently than less common host species in four avian communities in Africa, Asia and Europe.
To improve our understanding of avian malaria transmission, we sought to determine which biological factors (mosquito abundance and diversity; host ecological traits and density) explain the prevalence and diversity of Plasmodium lineages across seven locations along the Tapajós and Jamanxim rivers in Brazilian Amazonia. Specifically, we predicted that: (1) Plasmodium prevalence in a given host species would be positively correlated with the host species’ density; (2) Plasmodium lineage diversity would be positively correlated with avian host diversity; (3) avian ecological traits would explain some of the variation in Plasmodium prevalence; and (4) the prevalence and diversity of avian Plasmodium lineages would be positively correlated with the abundance and diversity of mosquitoes. A secondary motivation of our study was to provide information on the diversity and distribution of ornithophilic mosquitoes in a region of Amazonia that has not previously been explored with respect to these insects.
MATERIALS AND METHODS
Sampling sites
The study area was located midway down the Tapajós River, a major south bank tributary of the Amazon River. Sampling was carried out along both banks of the Tapajós River covering both the Rondônia and Tapajós areas of endemism (Silva et al. Reference Silva, Rylands and Fonseca2005). We also sampled the right bank of its most important tributary, the Jamanxim River (Fig. 1). The area comprises a wide variety of microhabitats, with the terra firme and igapó forests being the most broadly distributed. Sampling was conducted along six transects of five km in length, beginning in the seasonally flooded forest (igapó) and crossing the interior of terra firme forest. We also sampled a 250 m transect on a river island. We named these seven sites as follows: the first letter of the label is the first letter of the river name (‘T’ for Tapajós and ‘J’ for Jamanxim); the second letter is the margin of the river (‘L’ for left bank, ‘R’ for right bank and ‘I’ for the island); site names also include a number to identify unique sampling sites on the same bank of the same river.

Fig. 1. Map showing the seven sampling sites along the Tapajós and Jamanxim rivers. Background shading corresponds to elevation with lower areas represented by darker shading. The inset shows the state of Pará, Brazil, and the study area (SA) is represented as a dark grey rectangle within the state. Am, Amazon River; Jam, Jamanxim River; and Tap, Tapajós River.
Bird sampling
We placed five mist net lines within each transect. These five mist net lines were each separated by 1 km and contained ten nets. The 50 mist nets (12 m length × 3 m height) remained open for three consecutive mornings at each of the seven transect sites. Owing to the small size of the river island, the mist nets were arranged in a single 250 m line and were sampled with the same effort as the others sites. Blood sampling of captured birds took place during two distinct seasons, the dry season for the sites TL1, TL2, TL4, TR1 and JR1 (18 July–3 August 2012) and flooding period for the sites TL1, TL2, TL3 and TI (1–17 October 2012), thus only two sites, TL1 and TL2, were sampled during both the dry and flooding seasons. At each site, to allow sufficient time for transmission of Plasmodium to avian hosts, avian blood samples were collected from individual birds shortly after (~2–3 weeks) mosquito vectors were also sampled. Netted birds were bled by brachial venipuncture using heparinized capillary tubes. Blood samples were stored in 95% ethanol until DNA extraction. After blood collection, birds were ringed and released, or euthanized and prepared for museum specimens. All tissue samples and birds were collected or ringed under appropriate permits from Brazil (IBAMA no 22/2012 and ICMBio no 004/2012). Tissue samples and voucher specimens were deposited in the Bird Collection at the Instituto Nacional de Pesquisas da Amazônia – INPA, Manaus, Brazil. Species nomenclature follows Piacentini et al. (Reference Piacentini, Aleixo, Agne, Maurício, Pacheco, Bravo, Brito, Naka, Olmos, Posso, Silveira, Betini, Carrano, Franz, Lees, Lima, Pioli, Schunck, Amaral, Bencke, Cohn-Haft, Figueiredo, Straube and Cesari2015).
Besides mist net sampling, we conducted point count surveys (10 min each), spread along transects at every 500 m, totalling 11 points per transect. Each point was sampled for four consecutive days at each sampling period. We estimated density of individual species in each area using MCDS (Multiple Covariates Distance Sampling) as implemented in the program Distance 6.0 (Thomas et al. Reference Thomas, Laake, Rexstad, Strindberg, Marques, Buckland, Borchers, Anderson, Burnham, Burt, Hedley, Pollard, Bishop and Marques2009). The analyses were stratified to obtain density estimates for each area. We truncated 10% of data with larger distances within each species to avoid double counting the same individual, as recommended by Buckland et al. (Reference Buckland, Anderson, Burnham, Laake, Borchers and Thomas2001). For each species, we compared the following models: (1) half normal and hazard rate set as functions of expansion adjusted by cosine; (2) simple polynomial; and (3) polynomial hermite. We then calculated the Akaike's Information Criterion (AIC) for each model and chose the one with the lowest AIC score.
Mosquito sampling
Mosquitoes were collected using light traps powered by 12 V batteries as designed by Falcão (Reference Falcão1981). The traps were installed in the same sampling transects used for mist netting birds, and were distributed in two vertical layers, at ground level (suspended approximately 1 m above the ground) and in the forest canopy (suspended between 10 and 15 m above the ground). A total of 14 traps were installed simultaneously in each transect and these were located at equidistant points starting at the river bank and ending at 4 km from the river bank in the interior of terra firme forest. Mosquito collections were made during three consecutive nights, between 18:00 and 21:00 h during both the dry season for the sites TL1, TL2, TL4, TR1 and JR1 (2–13 July 2012) and the flooding season for the sites TL1, TL2, TL3 and TI (12–23 September 2012), thus only two sites, TL1 and TL2, were sampled during both the dry and flooding seasons. Mosquitoes were stored in small plastic vials labelled with the corresponding data for each sample. In the laboratory, the female mosquitoes were separated and identified taxonomically using external morphological characters observed using a stereoscope. We only used female mosquitoes for abundance estimation because only females are potential vectors for Plasmodium. For species determination, we used keys published by Galindo et al. (Reference Galindo, Blanton and Peyton1954) and Forattini (Reference Forattini1962, Reference Forattini1965a , Reference Forattini b , Reference Forattini2002). We used the infrageneric classification scheme of the genus Anopheles from McKeon et al. (Reference McKeon, Moreno, Sallum, Povoa and Conn2013), Moreno et al. (Reference Moreno, Bickersmith, Harlow, Hildebrandt, McKeon, Silva-do-Nascimento, Loaiza, Ruiz, Lourenço-de-Oliveira, Sallum, Bergo, Fritz, Wilkerson, Linton, Juri, Rangel, Póvoa, Gutiérrez-Builes, Correa and Conn2013) and Ruiz-Lopez et al. (Reference Ruiz-Lopez, Wilkerson, Ponsonby, Herrera, Sallum, Velez, Quiñones, Flores-Mendoza, Chadee, Alarcon, Alarcon-Ormasa and Linton2013), which differentiates among groupings of cryptic but presently unnamed lineages. The abbreviation of genera and subgenera follows the guidelines suggested by Reinert (Reference Reinert2001), according to the newly proposed mosquito nomenclature of the Walter Reed Biosystematics Unit, Smithsonian Institution (catalogue available at http://www.mosquitocatalog.org/taxon_table.aspx) and Harbach (Reference Harbach2013) in the Mosquito Taxonomic Inventory (www.mosquito-taxonomic-inventory.info/).
Parasite detection
DNA was extracted from avian blood samples using the Qiagen DNeasy 96 Blood and Tissue kit (Qiagen, Valencia, CA), following the Qiagen protocol for blood in 95% ethanol. Total DNA was screened by real-time PCR to detect haemosporidian DNA following the protocol of Bell et al. (Reference Bell, Weckstein, Fecchio and Tkach2015). Positive and negative controls were included in all real-time PCR runs. Samples identified as positive by real-time PCR underwent subsequent nested PCR, as outlined in Bell et al. (Reference Bell, Weckstein, Fecchio and Tkach2015), to amplify a 477 bp fragment of the cytochrome b gene.
Positive nested PCR products were purified using ExoSAP-IT (Affymetrix, Santa Clara, CA), sequenced using BigDye terminator v3.1 cycle sequencing kit (Applied Bio systems, Foster City, CA), and run on an ABI 3100 DNA sequencer (Applied Bio systems, Foster City, CA). For sequencing protocol and primers see Bell et al. (Reference Bell, Weckstein, Fecchio and Tkach2015). Forward and reverse sequences were visualized and assembled using Sequencher v.5.0.1 (Gene Codes Corp., Ann Arbor, MI). Chromatograms that showed the presence of multiple infections were scored as co-infections. Co-infections were separated using the program PHASE 2.1.1 (Stephens et al. Reference Stephens, Smith and Donnelly2001; Stephens and Donnelly, Reference Stephens and Donnelly2003) following the protocol of Harrigan et al. (Reference Harrigan, Sedano, Chasar, Nguyen, Whitaker and Smith2014). Assembled sequences were aligned using BioEdit v7.2.0 (Hall, Reference Hall1999) and collapsed to unique haplotypes using the FaBox haplotype collapser and converter tool (Villesen, Reference Villesen2007). Sequence identities were verified with a local BLAST against the MalAvi database (Bensch et al. Reference Bensch, Hellgren and Pérez-Tris2009) using BioEdit v7.2.0 (Hall, Reference Hall1999). New lineages were named after the host of origin following standard protocol (Bensch et al. Reference Bensch, Hellgren and Pérez-Tris2009), using a six-letter code produced by using the first three letters of both the host genus and specific epithet followed by a number to denote multiple lineages from a single host species. For example, lineage WILPOE01 represents the first lineage obtained from Willisornis poecilinotus. All sequences were deposited in GenBank (Accession No KU562250–KU562512) and the MalAvi database.
Assembled sequences of unique lineages were used to reconstruct a molecular phylogeny using Bayesian inference (BI) as implemented in MrBayes v. 3.2.2 (Huelsenbeck and Ronquist, Reference Huelsenbeck and Ronquist2001; Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003) and the GTR + I + G model of nucleotide substitution as determined by jModelTest (Darriba et al. Reference Darriba, Taboada, Doallo and Posada2012); Leucocytozoon fringillarum (FJ168564) served as the outgroup. The BI analysis was run until the s.d. of split frequencies stabilized below 0·01. Twenty-five percent of resulting trees were discarded as burn in. The resulting consensus tree was visualized in FigTree (Rambaut, Reference Rambaut2009).
Statistical analyses and modelling
We were interested in testing whether the prevalence of Plasmodium parasites was related to the density of avian hosts within the different sites sampled. To do this we first restricted our analysis to species sampled at least five times within a site; this left us with three sites (TL1, TL2 and TL3) each including more than three species that met our sample size criterion (Table A2). We then calculated prevalence for each species at each site separately as the number of Plasmodium-infected individuals divided by the total number of individuals sampled of a particular species at a particular site. Individuals infected with only Haemoproteus parasites were considered uninfected for this analysis since we did not sample the arthropod vectors for this parasite genus and because birds can be found with mixed infections of both genera (Valkiūnas, Reference Valkiūnas2005). Individual birds infected with both Plasmodium and Haemoproteus were considered infected. We then ran three generalized linear models, one for each site, with prevalence as the response variable and host density as the explanatory variable with a quasibinomial error structure to account for overdispersion (Bolker et al. Reference Bolker, Brooks, Clark, Geange, Poulsen, Stevens and White2009). We entered two vectors of infected and uninfected individuals into the model to weight prevalence by sample size as is standard when running such models in R; see Crawley, Reference Crawley2012). Since sampling took place in two seasons for sites TL1 and TL2, we compared prevalence of Plasmodium parasites between seasons (within sites; prevalence was calculated for all individuals, i.e. not separated by host species, sampled within each season for each of the two sites) using chi-squared tests to confirm that seasons could be grouped for this analysis.
We tested the hypothesis that Plasmodium lineage diversity was related to the diversity of avian hosts across all seven sampling sites. For this we calculated a Simpson's index of diversity for all avian hosts that were sampled (117 species) and one for all parasite lineages within each sampling site using the ‘diversity’ function in the R package vegan (Oksanen et al. Reference Oksanen, Blanchet, Kindt, Legendre, Minchin, O'Hara, Simpson, Solymos, Stevens and Wagner2015). We then used Spearman's rank correlation tests (correlation statistic is ρ) to determine whether the variables were correlated across sampling sites.
We were also interested in testing whether the prevalence of Plasmodium parasites across avian host species was related to the following host ecological variables: nest location (ground, understory, sub canopy, canopy and cliff/bank), nest type (open cup, closed cup and cavity), flocking behaviour (solitary/family, single species and mixed species), and diet (insectivore, frugivore/granivore and omnivore). We scored these traits for all species sampled using a combination of The Birds of South America Volumes I and II (Ridgely and Tudor, Reference Ridgely and Tudor1989a , Reference Ridgely and Tudor b ), Neotropical Birds: Ecology and Conservation (Stotz et al. Reference Stotz, Fitzpatrick, Parker and Moskovits1996), The Cornell Lab of Ornithology: Neotropical Birds (http://www.neotropical.birds.cornell.edu/portal/home) and WikiAves (http://www.wikiaves.com.br). We pooled our site data for this analysis because: (1) we did not expect site to influence the relationship of prevalence and ecological host traits (e.g. Matthews et al. Reference Matthews, Ellis, Hanson, Roberts, Ricklefs and Collins2016); and (2) because we did not find any significant differences in the prevalences of individual species sampled at multiple sites in our study (results not shown). We therefore constructed a generalized linear model with Plasmodium prevalence (weighted by sample size) as the response variable and each of the ecological variables as explanatory variables. We ran the model with a quasibinomial error structure to account for overdispersion and only included species with at least five individuals sampled (n = 44 avian species). Initially we included the taxonomic family of host species as an explanatory variable in the model to account for potential differences in prevalence among families, but sparse sampling led to uninterpretable estimates of family-level prevalences and so we dropped family from our final model. We also ran Wald chi-squared tests on each of the explanatory variables in the model using the function ‘wald.test’ in the R package aod (Lesnoff and Lancelot, Reference Lesnoff and Lancelot2012).
Finally, we tested the hypothesis that the prevalence and diversity of Plasmodium lineages were positively related to the abundance and diversity of mosquitoes across all seven of our sampling sites using Spearman's rank correlation tests. Here we calculated parasite prevalence for sites rather than for species by dividing the total number of infected individuals by the total number of individuals sampled in a site irrespective of host species. We again calculated a Simpson's index of diversity for Plasmodium lineages and one for mosquitoes within each sampling site.
All statistical analyses were performed in R version 3.2.3 (R Core Team, 2015).
RESULTS
Prevalence and diversity of Plasmodium lineages
We analysed 675 birds of 120 species sampled in seven communities along the Tapajós and Jamanxim rivers (Fig. 1, Table A1). Plasmodium infections were detected in 136 individuals from 51 host species with a prevalence of 20% (Table A1). Infection prevalence varied among well sampled host species (>10 individuals screened), ranging from 0 to 64% (Table A1). Plasmodium prevalence was homogeneous across sites, ranging from 12 to 31% (χ 2 = 11·282, d.f. = 6, P = 0·080; Table 1). Based on cytochrome b divergence, we recovered 89 haemosporidian lineages within the genus Plasmodium, of which 81 (91%) were reported for the first time (Fig. 2). Although there are several well resolved and supported clades within the phylogeny, the general pattern is one of many polytomies with low node support (Fig. 2). When mapped onto the phylogeny, host family shows little perceivable pattern within the phylogeny, dominated by the highly sampled family Thamnophilidae. In several cases, individual Plasmodium lineages were recovered from more than one host family, with three Plasmodium lineages found in individual hosts from four different host families.

Fig. 2. BI phylogenetic reconstruction of Plasmodium lineages recovered from sites along the Tapajόs and Jamanxim rivers. Posterior probability support above 0·9 is noted at the base of nodes and host family is noted next to terminal taxon labels.
Table 1. Prevalence of Plasmodium per site along the Tapajós and Jamanxin rivers, southeastern Amazonia, Brazil. Specific site location information can be found in Fig. 1

We sampled birds at sites TL1 and TL2 during the dry and flooding periods, allowing us to test for seasonal differences in prevalence. We found no differences in overall prevalence of Plasmodium parasites between the two seasons within each site (site TL1, χ 2 = 0, d.f. = 1, P = 1; site TL2, χ 2 = 0·703, d.f. = 1, P = 0·402). For these tests we used ten host species from site TL1 and nine host species from the site TL2 for which we had sampled at least five individuals. We also found no differences in prevalence between seasons for individual host species (results not reported).
Avian host ecology and Plasmodium prevalence
Prevalence of Plasmodium (calculated for each host species within sites) was positively related to host density at site TL3 (GLM coefficient = 0·186 ± 0·05 s.e., P = 0·037, n = 5), but not at sites TL1 (0·027 ± 0·09 s.e., P = 0·769, n = 8) and TL2 (−0·096 ± 0·20 s.e., P = 0·661, n = 6). However, the relationship at site TL3 was based on only five host species, and would not be considered significant after a Bonferroni correction (after three tests, alpha = 0·05/3 or 0·017; Table A2). The diversity of Plasmodium parasites was not related to the diversity of avian hosts (ρ = 0·49, P = 0·268, n = 7).
We found no relationship between ecological traits of the hosts and the prevalence of Plasmodium parasites. We report coefficients and significance of each category of the explanatory ecological variables in Table 2 (results of each host trait modelled separately can be found in Table A3). We also ran Wald chi-squared tests for each ecological variable across all coefficients and none were significant (nest location: χ 2 = 1·4, d.f. = 4, P = 0·85; nest type: χ 2 = 4·6, d.f. = 2, P = 0·10; flocking behaviour: χ 2 = 1·6, d.f. = 2, P = 0·45; diet: χ 2 = 2·1, d.f. = 2, P = 0·35). For this analysis, we used 504 birds of 44 species with a minimum of five individuals sampled per species. Plasmodium infections were detected in 101 individuals from 23 host species with a prevalence of 20% (Table A4).
Table 2. The results of a generalized linear model relating four avian ecological traits to the prevalence of Plasmodium parasites in avian hosts

All of the explanatory ecological variables were categorical. We therefore report the estimate of the coefficient of each of the levels of those variables in relation to a base level. The base levels are as follows: nest location is ground, nest type is open cup, flocking is solitary/family, diet is insectivore. We report the estimate of the coefficient for each variable, its s.e., t value and P value; the null deviance of the model is 198·01 on 43 d.f. and the residual deviance is 128·35 on 33 d.f.
Mosquitos, prevalence and diversity of Plasmodium lineages
We collected 511 female mosquitoes from 56 species and morpho-species belonging to 11 genera (Tables A5 and A6). We ran paired Mann–Whitney U tests to compare mosquito abundance between seasons in sites TL1 and TL2. In site TL1, mosquito abundance was higher in the flooding period than in the dry season (U = 21·5, P < 0·001), but in site TL2, there was no difference in mosquito abundance between seasons (U = 169, P = 0·441). In the other five sites, mosquito collection took place in only one season, preventing further seasonal comparisons (Tables A5 and A6).
The prevalence of Plasmodium parasites in avian hosts (calculated for an entire site and not by host species) was not related to the diversity (ρ = −0·46, P = 0·302) or the abundance (ρ = −0·22, P = 0·641) of mosquitoes across the seven sampling sites. The diversity of Plasmodium parasites was also not related to the diversity of mosquitoes (ρ = 0·67, P = 0·102), but was positively related to the abundance of mosquitoes (ρ = 0·79, P = 0·034) across sampling sites (Fig. 3). Since each of these tests represents a separate hypothesis, we did not apply a Bonferroni correction to the resulting P values.
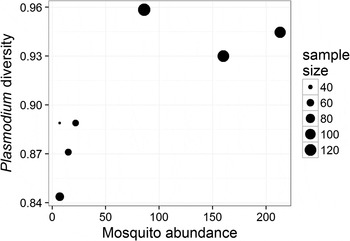
Fig. 3. Relationship between the diversity of Plasmodium parasites (calculated using Simpson's index of diversity) and mosquito abundance at each of the sampled sites; point size is scaled to the number of individual birds sampled at each site. The two variables are positively correlated (ρ = 0·79, P = 0·034).
DISCUSSION
We investigated biotic factors that may determine the prevalence and diversity of Plasmodium parasites in Amazonian birds. We found that the diversity of Plasmodium lineages was positively correlated with mosquito abundance across the seven bird communities we sampled. Neither bird density nor bird diversity explained prevalence or lineage diversity of Plasmodium parasites among avian hosts. None of the four ecological traits of avian hosts explained Plasmodium prevalence. The lack of seasonal differences in prevalence of Plasmodium found along the Tapajós River needs to be considered with caution since we tested this with samples collected from only two communities.
Our finding that Plasmodium lineage diversity is correlated with mosquito abundance and not with any avian host traits suggests that diversity and distribution of these parasites might be constrained by the final host (Culicidae) in our bird–parasite–mosquito system. Ishtiaq et al. (Reference Ishtiaq, Guillaumot, Clegg, Phillimore, Black, Owens, Mundy and Sheldon2008) demonstrated that the movement of Plasmodium lineages among southwest Pacific Islands might be restricted by the lack of overlap in the distributions of competent vector species. One possibility is that the mosquito community in our study region only has a few competent mosquito vectors and their signal was washed out by the huge mosquito diversity found in southeastern Amazonia. Alternatively, regional processes at the level of host populations, such as immunity to particular parasite lineages or differential exposure to certain parasite lineages, might mask any density-dependent influence of avian hosts and mosquitoes vectors on prevalence. For instance, the distribution and diversity of many parasites among host populations are known to be highly variable and one of the main reasons behind this is the inequality of individual host immune responses in defending themselves against particular parasites (Poulin, Reference Poulin2007).
Our results demonstrate that Plasmodium lineage diversity and prevalence in Amazonian birds does not vary with host ecological traits and avian host density. We expected that host population density would be positively correlated to avian malaria prevalence since it affects vector–host-encounter rates (Dobson, Reference Dobson2004). Although several studies have found evidence for higher haemosporidian prevalence in denser host populations (Matthews et al. Reference Matthews, Ellis, Hanson, Roberts, Ricklefs and Collins2016; Ricklefs et al. Reference Ricklefs, Soares, Ellis and Latta2016; Ellis et al. Reference Ellis, Medeiros, Collins, Sari, Coffey, Dickerson, Lugarini, Stratford, Henry, Merrill, Matthews, Hanson, Roberts, Joyce, Kunkel and Ricklefs2017), many others failed to find such an association (Svensson-Coelho et al. Reference Svensson-Coelho, Blake, Loiselle, Penrose, Parker and Ricklefs2013; Gonzalez-Quevedo et al. Reference Gonzalez-Quevedo, Davies and Richardson2014). Kilpatrick et al. (Reference Kilpatrick, Daszak, Jones, Marra and Kramer2006) showed that West Nile virus transmission within a local host community was influenced by extreme heterogeneity in mosquito feeding patterns. At least in some sites, transmission of multi-host pathogens such as avian malaria, may be influenced by heterogeneity in host–vector compatibility more than by bird density (Kilpatrick et al. Reference Kilpatrick, Daszak, Jones, Marra and Kramer2006; Medeiros et al. Reference Medeiros, Hamer and Ricklefs2013). Specifically, for avian malaria, higher abundance of vectors does not lead to a higher host–vector-encounter rate. For example, Medeiros et al. (Reference Medeiros, Ricklefs, Brawn and Hamer2015) showed that vectors overutilized some bird species regardless of their abundance and Plasmodium prevalence may be associated with vector utilization rather than vector abundance.
Nest characteristics among many other ecological traits of host individuals might be associated with variation in haemosporidian prevalence (Ricklefs et al. Reference Ricklefs, Swanson, Fallon, Martinez-Abrain, Scheuerlein, Gray and Latta2005; Fecchio et al. Reference Fecchio, Lima, Silveira, Braga and Marini2011, Reference Fecchio, Lima, Svensson-Coelho, Marini and Ricklefs2013; Svensson-Coelho et al. Reference Svensson-Coelho, Blake, Loiselle, Penrose, Parker and Ricklefs2013; González et al. Reference González, Matta, Ellis, Miller, Ricklefs and Gutiérrez2014; Lutz et al. Reference Lutz, Hochachka, Engel, Bell, Tkach, Bates, Hackett and Weckstein2015; Matthews et al. Reference Matthews, Ellis, Hanson, Roberts, Ricklefs and Collins2016). However, based on simple correlations, these studies relied on the premise that nestlings or adults are more exposed to the vectors in the nest according to its architecture. Moreover, mixed results found in these studies suggest that the relationship between nest type and risk of infection by haemosporidian parasites might be location dependent. An analysis of the identity of host blood found in engorged female mosquitoes could provide a general test for this pattern and confirm whether host nest type can predict haemosporidian prevalence in birds.
Lack of seasonal variation in Plasmodium prevalence found in two communities sampled along the Tapajós River confirms the temporal stability of these parasites in tropical birds. There are three main possible explanations for this: (1) the abundance of mosquito vectors might differ between seasons (i.e. site TL1), but even the lower number of actively feeding mosquitoes is sufficient to ensure transmission especially if these few mosquitoes are the most competent vector for more prevalent Plasmodium lineages; (2) infections in birds last long enough to span more than a single season and thus mask the changes in mosquito abundance between seasons; and (3) vector abundance might be stable over seasons (i.e. site TL2), allowing Plasmodium transmission throughout the year. If Plasmodium parasites have a dynamic aspect in tropical bird communities, it may vary with years or decades and not between seasons within the same year.
Several species of mosquito belonging to the genera Aedeomyia, Anopheles, Coquillettidia, Culex, Culiseta, Mansonia, Aedes (Ochlerotatus and Stegomyia) have been implicated in the transmission of Plasmodium spp. in birds (Valkiūnas, Reference Valkiūnas2005; Njabo et al. Reference Njabo, Cornel, Sehgal, Loiseau, Buermann, Harrigan, Pollinger, Valkiūnas and Smith2009). Among the species of mosquito inhabiting our study area in Tapajos, Ad. (Ady.) squamipennis is known as a natural vector of avian malaria in Venezuela (Gabaldon et al. Reference Gabaldon, Ulloa and Pulido1981). Another Neotropical mosquito Culex (Melanoconion) ocossa, together with Ad. (Ady.) squamipennis could be responsible for the transmission of avian malaria in some regions of Panama (Loaiza and Miller, Reference Loaiza and Miller2013). Unfortunately, we were not able to analyse the engorged females of these mosquitoes to study vectorial capacity or host specificity and thus future research on this is necessary as a first step to fully understand the transmission risk and high diversity of avian malaria parasites in the Neotropical region.
The absence of any perceivable pattern of host family within the phylogeny can be attributed to both the high level of sampling from the host family Thamnophilidae and the low level of host specificity known for Plasmodium (Beadell et al. Reference Beadell, Gering, Austin, Dumbacher, Peirce, Pratt, Atkinson and Fleischer2004, Reference Beadell, Covas, Gebhard, Ishtiaq, Melo, Schmidt, Perkins, Graves and Fleischer2009; Valkiūnas, Reference Valkiūnas2005; Dimitrov et al. Reference Dimitrov, Zehtindjiev and Bensch2010; Ishtiaq et al. Reference Ishtiaq, Clegg, Phillimore, Black, Owens and Sheldon2010). This low level of host specificity is shown in those lineages recovered from three and four different host families, spanning different host orders. Host switching is an important evolutionary mechanism in avian haemosporidian parasites with closely related haemosporidian lineages conserved within higher host taxa (Waldenström et al. Reference Waldenström, Bensch, Kiboi, Hasselquist and Ottoson2002; Križanauskiené et al. Reference Križanauskiené, Hellgren, Kosarev, Sokolov, Bensch and Valkiūnas2006; Ricklefs et al. Reference Ricklefs, Outlaw, Svensson-Coelho, Medeiros, Ellis and Latta2014). Due to high levels of host switching and subsequent dispersal, cospeciation is not thought to have played a large role in the evolutionary history of Plasmodium (Ricklefs et al. Reference Ricklefs, Outlaw, Svensson-Coelho, Medeiros, Ellis and Latta2014; Lauron et al. Reference Lauron, Loiseau, Bowie, Spicer, Smith, Melo and Sehgal2015). However, the high host diversity in Amazonia would be an ideal system to explore the existence of coevolutionary links between Plasmodium parasites and avian hosts in future studies.
The heterogeneous prevalence of Plasmodium across bird species in southeastern Amazonia, regardless of their ecological traits, suggests that constraints on the distribution of these parasites are related to vectors within these assemblages. This highlights the importance of exposure to vectors in explaining avian malaria prevalence (Medeiros et al. Reference Medeiros, Ricklefs, Brawn and Hamer2015). Nonetheless, the results have to be interpreted cautiously, because of low sample sizes within sites and the low number of sites in general. Therefore, the next step in understanding the factors promoting the high diversity and heterogeneity of Plasmodium lineages in this region of Amazonia, as well as the mechanisms that produce variation in the prevalence of these vector-borne parasites across avian hosts, must include studies that integrate rates of vector exposure, feeding preference and vectorial capacity of the mosquitoes in the same area.
ACKNOWLEDGEMENTS
We thank the ornithologists: Marcelo Barreiros, Gabriel Leite, Thiago Bicudo, Thiago Moura, Daniel Gressler, Camila Duarte and several Munduruku Indians who helped collect the blood samples. We also thank Camila Ribas and Mario Cohn-Haft from the Bird and Genetic Resources Collections of Instituto Nacional de Pesquisas da Amazônia who loaned the samples used in this study. We also thank three anonymous referees for their comments on earlier versions of the manuscript and Vitor Piacentini who kindly reviewed the bird nomenclature.
FINANCIAL SUPPORT
This work was funded by the Fundação de Amparo à Pesquisa do Estado do Amazonas – FAPEAM and Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (Process number 61.0012/2008.7-DCR/CNPq/FAPEAM) to A.F. and US National Science Foundation grants DEB-1503804 to J.D.W. and DEB-1120734 to V.V.T. During the project, A.F. was supported by a postdoctoral fellowship from CNPq (Process numbers 350140/20120 and 201275/2014-7) and he is currently maintained by a PNPD scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Appendix
Table A1. Plasmodium prevalence from seven bird communities in southeastern Amazonia, Brazil. Species of birds are organized taxonomically

Table A2. The 11 bird species used to test the effect of host density (individuals per 100 ha) on prevalence of Plasmodium from three transects along the Tapajós River. The geographic locations of sites can be found in Fig. 1

Table A3. In the main text of our paper we report the results of a multiple regression (GLM) modelling prevalence as a function of four explanatory variables (nest location, nest type, flocking and diet). Here we report the results of four separate models of each explanatory variable by itself, for comparison. Each model is a GLM with a ‘quasibinomial’ error structure (to account for overdispersion as in the model presented in the main text); coefficients are reported as in Table 2 of the main text

Table A4. The 44 species used in the analysis of an effect of avian ecological traits on Plasmodium prevalence in southeastern Amazonia
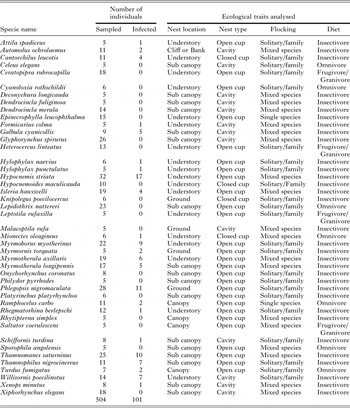
Table A5. Mosquito species caught across five sites during the dry season. The geographic locations of sites can be found in Fig. 1

Table A6. Mosquito species caught across four sites during the flooding season. The geographic locations of sites can be found in Fig. 1












