


Hereditary spastic paraplegia (HSP) is a clinically and genetically heterogeneous disorder that involves progressive weakness and spasticity of the lower extremities followed by a similar decline in the upper extremities.Reference Salinas, Proukakis, Crosby and Warner 1 Clinically, HSP is divided into two forms: the pure form variant demonstrates spasticity as the major presenting feature, whereas the complex variant exhibits additional symptoms including ataxia, mental retardation, peripheral neuropathy, muscle atrophy, and/or dysarthria.Reference Fink 2 , Reference Harding 3 Genetically, HSP can also be classified according to the pattern of inheritance, which can be autosomal recessive, autosomal dominant, X-linked recessive, and sporadic. Autosomal recessive HSP with thin corpus callosum (ARHSP-TCC) is one form of this disease, and alterations of the SPG 11 gene account for 41% to 77% of ARHSP-TCC cases.Reference McDermott, White, Bushby and Shaw 4 , Reference Paisan-Ruiz, Dogu, Yilmaz, Houlden and Singleton 5 In the current study, members of a Chinese HSP pedigree were clinically and genetically examined to characterize the phenotype and to identify the genotype. We identified a novel compound heterozygous mutation in the SPG 11 gene by using a combined approach that included targeted exome capture technology and candidate mutation validation.
Materials and Methods
Ethics Statement
The use of clinical information and human-derived materials in this study was approved by the Ethical Committee of Xuan Wu Hospital, Capital Medical University. Written informed consent was obtained from all subjects or from the parents or legal guardians of minor subjects.
Subjects
The pedigree chart is illustrated in Figure 1. A detailed medical history was obtained from nine individuals, including two patients (II-2, II-4) and seven family members (I-1, I-2, II-1, II-6, III-1, III-2, and III-3). All of these individuals were physically examined by two experienced neurologists (MW, XZ) and underwent genetic testing. The proband was a young man (II-4), and the other patient was his older sister (II-2). Other family members (including the patients’ parents, siblings, and children) had normal clinical presentations. The patients were not the products of a consanguineous relationship. Detailed information regarding the age of onset, the progression of disability, and the clinical manifestation was obtained. Mental impairment was measured by the Wechsler Adult Intelligence Scale-Revised in China (WAIS-RC). Magnetic resonance imaging (MRI), electromyogram (EMG) and cerebrospinal fluid (CSF) analysis were performed for both patients.
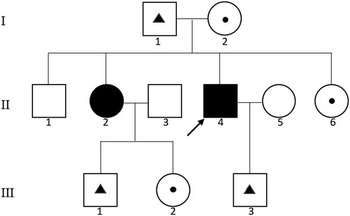
Figure 1 Pedigree of the family. Squares indicate males; circles, females; arrow, proband; solid symbols, affected individual; small triangle with square or circle, c.4001_4002insATAAC mutation carrier; small dot with square or circle, c.4057C>G mutation carrier.
Mutation Analysis
Screening for Mutations
Blood samples were collected from all nine family members. We then extracted genomic DNA according to the manufacturer’s instructions (D2492 Blood DNA Maxi Kit, Omega Bio-Tek, Norcross, GA). Based on targeted exome capture technology, a specific HSP panel was generated and used to collect the protein coding regions of 56 targeted genes (Table 1). Next, we prepared exon-enriched DNA libraries for high-throughput sequencing using the Illumina HiSeq 2000 platform. Initially, targeted exome sequencing was performed on the proband. A mean exome coverage of more than 98.1% was obtained, with a variant accuracy of more than 95%. These changes were computationally filtered against exome data from ethnic Han Chinese in Beijing, available through the 1000 Genomes Project (http://www.1000genome.org), against the Han Chinese Beijing SNPs in dbSNP131, the National Heart, Lung, and Blood Institute GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), and the data from 5,038 in-house subjects (from the Beijing Genomics Institute, Shenzhen, China).
Table 1 HSP-associated genes included in the clinical screening panel
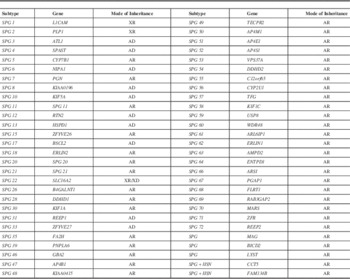
AD=autosomal dominant inheritance; AR=autosomal recessive inheritance; XD=X chromosome dominant inheritance; XR=X chromosome recessive inheritance.
Mutation Validation
The variants identified in the proband were confirmed by direct polymerase chain reaction product sequencing using Bigdye terminator V3.1 cycle sequencing kits (Applied Biosystems, Foster City, CA) and analyzed on an ABI 3130XL genetic analyzer. Whether the variants that were detected by targeted exome sequencing co-segregated with the disease phenotype in the family was tested by Sanger sequencing. Primer pairs were designed using a primer program (http://www.yeastgenome.org/cgi-bin/web-primer) (DNA reference number NG_009759) (Table 2).
Table 2 The sequence of PCR primers and expected PCR product size

Results
Clinical Data
The proband (II-4) was a 29-year-old male who suffered from progressive weakness and stiffness of the lower extremities for 5 years and experienced a similar decline in the upper extremities for 1 year. By the time of hospitalization, the patient walked with scissors gait. His older sister (II-2) was 32 years old and had developed progressive weakness of the lower extremities and gait disturbance 20 years ago, accompanied with urinary incontinence for more than 14 years. She had been wheelchair-bound for 4 years by 2014. Notably, their school performances were worse than those of their unaffected siblings and classmates, and this difference showed much earlier than the motor dysfunction. Unfortunately, because of the parents’ poor awareness of mental retardation in their children, they did not noticed their children had poor intelligence performance until they entered a privilege school, which was at approximately 6 years of age. The full-scale IQ scores of patients II-4 and II-2 examined during hospitalization were 75 and 53, respectively. However, no ophthalmoplegia, decreased vision, dysphagia, cerebellar signs, or extrapyramidal symptoms were observed in either patient. There were an additional two siblings (II-1 and II-6) in the parents’ generation. The patients’ parents (I-1 and I-2) were both 55-year-old farmers. Children III-1 and III-2 of patient II-2 and child III-3 of patient II-4 were both 8 years of age and performed well in school and extracurricular activities including athletics. All seven of these individuals were unaffected and had no complaints of neurological symptoms.
Neurological examination of patients II-4 and II-2 revealed weakness and spasticity of the lower extremities with positive pyramidal signs and pes cavus deformity. Additionally, II-4 exhibited additional upper extremities weakness, whereas II-2 suffered from dysarthria. No sensory impairment or hyperpigmentation of the skin was observed in either patient. Examination of the brain MRI scan revealed that both patients possessed a TCC and periventricular leukoaraiosis (Figure 2). An EMG was performed on each of the patients and revealed the presence of underlying peripheral nerve damage in II-2. Cerebrospinal fluid results were normal for both patients. Detailed information on the two patients is summarized in Table 3. Both patients were classified as having complicated HSP.
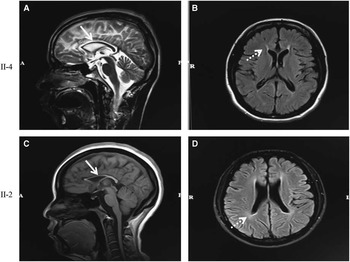
Figure 2 MRI features of the brain in patients II-4 and II-2. Both patients showed thinning of the corpus callosum on sagittal T2-weighted imaging (A) and sagittal T1-weighted imaging (C) images (arrow). Periventricular leukoaraiosis was found symmetrically in the anterior and posterior horn on fluid-attenuated inversion recovery imaging (B and D, dotted arrow).
Table 3 Clinical features of affected individuals in the family
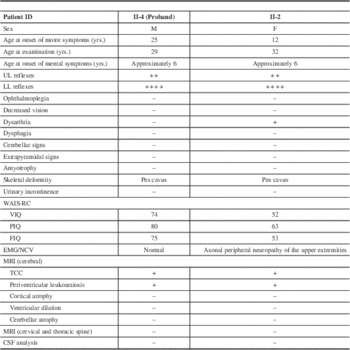
M=male; F=female; UL=upper extremities; LL=lower extremities; WAIS-RC=Wechsler Adult Intelligence Scale-Revised in China; VIQ=Vocable Intelligence Quotient; PIQ=Performance Intelligence Quotient; FIQ=Full Intelligence Quotient; MCV=motor nerve conduction velocity; NCV=nerve conduction velocity; CSF=cerebral spinal fluid; TCC=thin corpus callosum; +, presence; –, absence.
Mutation Analysis of the SPG 11 Gene
Using next-generation targeted sequencing, we found two novel compound heterozygous mutations (c.4001_4002ins ATAAC and c.4057C>G) in the SPG 11 gene in the proband (Figure 3A). Because neither mutation has been previously described, sequencing was extended to validate the identified variants (Figure 3B). Sanger sequencing demonstrated that these compound heterozygous mutations in SPG 11 were co-segregated with the disease phenotype described by the family pedigree. The detailed results of mutation in this family are illustrated in Figure 1. The affected sister (II-2) also possessed both of these mutations (Figure 4). Individuals who carried either heterozygous mutation were asymptomatic. The unaffected father (I-1), the son of the proband (III-3), and the son of the female patient (III-1) carried the insertion mutation (c.4001_4002insATAAC) in the heterozygous state. The patients’ unaffected mother (I-2), the unaffected youngest sister (II-6), and the daughter of the female patient (III-2) carried the missense heterozygous mutation (c.4057C>G). This mode of inheritance is consistent with an autosomal recessive pattern. The c.4001_4002insATAAC mutation in exon 24 was inherited from the healthy father; this mutation leads to a reading frame shift during transcription, resulting in premature termination of the protein product. The missense mutation c.4057C>G (p.H1353D) was inherited from the unaffected mother; this mutation causes an amino acid substitution from histidine to aspartic acid. It is predicted to be probably damaging by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) with a score of 0.99 in HumVar (Figure 5), and predicted to affect protein function by SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html) with a score of 0.00. In addition, conservation analysis of protein revealed that the substituted amino acid is highly conserved among different species (Figure 6); therefore, both mutations are likely to be pathological. Based on the results described here, we propose that these novel compound heterozygous mutations in SPG 11, c.4001_4002insATAAC and c.4057C>G, are the genetic cause of ARHSP in this Chinese family.
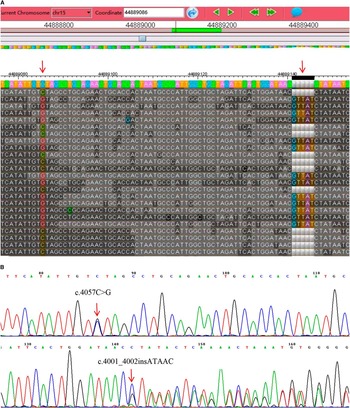
Figure 3 Identification of the mutations. Variants were detected by targeted exome sequencing (A) and confirmed by Sanger sequencing (B).

Figure 4 Sanger sequencing results of the female patient II-2. Sanger sequencing confirmed that II-2 possessed the same mutations as the proband.
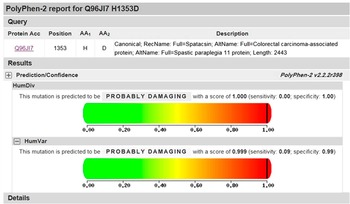
Figure 5 PolyPhen-2 analysis for c.4057C>G. The mutation of c.4057C>G was predicted to be probably damaging with a score of 1.00 in HumDiv and 0.99 in HumVar.

Figure 6 Conservation analysis. The protein sequence alignment demonstrates that H1353 is highly conserved among all of the species examined.
Discussion
We identified two novel SPG 11 gene mutations: an insertion mutation (c.4001_4002insATAAC) and a missense mutation (c.4057C>G). These mutations were found in a Chinese ARHSP-TCC pedigree.
To date, 15 distinct loci associated with HSP-TCC have been identified: SPG 1, SPG 11, SPG 15, SPG 18, SPG 21, SPG 44, SPG 45 (65), SPG 46, SPG 47, SPG 49, SPG 54, SPG 56, SPG 63, SPG 67, and SPG 71.Reference Boukhris, Feki and Elleuch 6 , Reference Finsterer, Loscher, Quasthoff, Wanschitz, Auer-Grumbach and Stevanin 7 It is difficult to determine the disease-causing gene based on the clinical manifestations because a number of coding exons exist in these genes, and traditional screening for each gene is infeasible for clinical applications. We therefore targeted a subset of genes that were potentially responsible for HSP. As a result, we demonstrated that an approach based on exome sequencing of known causative target genes of HSP is an efficient way of identifying pathological genes of interest.
The human SPG 11 gene, which is located at chromosome 15q13-15, encodes the protein SPATACSIN; this protein is prominently expressed in the cerebellum, cerebral cortex, hippocampus, and pineal gland.Reference Samaranch, Riverol and Masdeu 8 It is believed that HSP is a length-dependent distal axonopathy of the corticospinal tracts, resulting in lower limb spasticity and weakness. Several studies are working on alterations in the shaping of organelles, particularly the endoplasmic reticulum, as well as intracellular membrane trafficking and distribution as primary defects underlying the HSP.Reference Blackstone 9 – Reference Renvoise, Chang and Singh 11 Even so, the protein is still of a lot of unknown characteristics. Among the mutations that have been described in HSP, frame shift, or nonsense mutations are the most frequent types reported in SPG 11.Reference Cao, Rong and Huang 12 Building on this current knowledge base, our study identified two additional mutations, one insertion mutation and one missense mutation, in the SPG 11 gene. To some extent, these newly identified variants broaden the spectrum of SPG 11 mutations in Chinese patients and may warrant further investigation in patients.
This family included two affected patients, both of whom presented with weakness and spasticity of the extremities, mental impairment, and a TCC. The disease was absent in the parents and parental consanguinity was not present. Therefore, an autosomal recessive mode of inheritance is almost certainly the case considering that the patients suffered from a compound SPG 11 genotype. Together, these clinical observations are consistent with the diagnosis of HSP-TCC.Reference Shibasaki, Tanaka and Iwabuchi 13 , Reference Casali, Valente and Bertini 14
ARHSP caused by SPG 11 mutations is characterized by early-onset progressive spasticity and weakness, a TCC, and cognitive deficits. Approximately 79% of HSP-TCC patients first present with difficulty walking, and only 16% of patients initially present with signs of mental retardation.Reference Stevanin, Azzedine and Denora 15 Our patients showed prominent intellectual disability. This was reflected not only in a low IQ, but also in the onset time of mental retardation, which was earlier than the onset of the abnormal gait. Other clinical manifestations in these patients included spastic paraplegia of the lower limbs, and pes cavus deformity. Patient II-2 also exhibited urinary incontinence, dysarthria and hyperreflexia of the upper limbs. All of these manifestations have been reported in previous studies.
The two patients in the current study did not develop identical symptoms even though they carried the same genetic mutations. In this case, the differences can be interpreted as being as important as the similarities. The female patient started displaying symptoms at age 12, and the disease progressed over the course of 20 years. However, the proband began to develop similar symptoms much later, at the age of 25, and his disease progressed much more quickly. The female patient also showed obvious urinary incontinence, which was notably absent in her brother, who was also a patient. Additionally, the EMG results revealed peripheral neuropathy only in the sister (although no overt symptoms were reported by the patient). We inferred that the peripheral neuropathy was chronic and subclinical. Both patients in this study showed cerebral white matter changes on standard MRI (Figure 2), a finding that was recently reported.Reference Casali, Valente and Bertini 14 , Reference Lossos, Stevanin and Meiner 16 – Reference Ma, Xiong and Chang 19 The patients exhibited slight differences in their MRI results; the proband displayed remarkable changes in the anterior region, whereas the sister demonstrated alterations primarily in the posterior aspect. The reason for these differences in the clinical manifestation of the two sibling patients is currently unclear, but it is important to consider several points. First, it is well known that HSP is heterogeneous with regard to its clinical and genetic aspects. Second, the duration of the proband’s disease was much shorter than the sister’s. The possibility that the proband will develop new symptoms or that the present symptoms will worsen in the future cannot be excluded. Regular follow-up evaluations are scheduled to further describe the evolution of this patient’s disease.
In summary, the present results demonstrate that novel compound heterozygous mutations in SPG 11 are associated with ARHSP in this Chinese pedigree. The results contribute to understanding of the SPG 11 gene mutation spectrum and highlight the importance of genetic testing in HSP-TCC. Further studies are warranted to investigate the concrete genetic pathogenic mechanisms of HSP-TCC.
Acknowledgments
The authors acknowledge all of the patients and healthy subjects for their participation in this study.
Disclosures
Xiaojie Tian, Min Wang, Kaiyuan Zhang, and Xinqing Zhang do not have anything to disclose.
Statement of Authorship
XT and MW contributed equally to this work.









