


A 68-year-old man presented with a bilateral tonic-clonic seizure. His medical history included migraine with aura, hypertension and dyslipidemia. His father and mother had ischemic strokes in their fifth decade of life. On initial examination, his vitals were stable, but he had post-ictal confusion, transient right homonymous hemianopsia and mild right-sided Todd’s paresis. General systems examinations were unremarkable. MRI of the head showed multifocal confluent white matter lesions (Figures 1A-1B) with a few short association (U) fibers abutting the cortex, notable in the left occipital lobe (Figure 1C). An electroencephalography (EEG) captured a focal seizure originating from the left occipital lobe. Lumbar puncture revealed elevated protein (864 mg/L) without pleocytosis. Serum metabolic, infective and inflammatory workups were unremarkable. Levetiracetam 20 mg/kg was loaded intravenously to control his seizures. Subsequent neurological examination demonstrated resolution of his deficits. On further history taking, it was found that the patient had been diagnosed with migraine with aura since the age of 9 years. He described his headache to be bi-frontal, of moderate to severe intensity, throbbing in nature with associated photophobia, phonophobia and nausea. He was experiencing ~4 attacks/year with good relief from taking sumatriptan 100 mg. In some occasions, his headache would be preceded by a visual aura: he would experience either a gradual- or acute-onset right visual field cut followed by a headache. His aura could last between 1 and 4 hours. Six months before presentation, he started to experience frequent isolated visual auras without headaches.
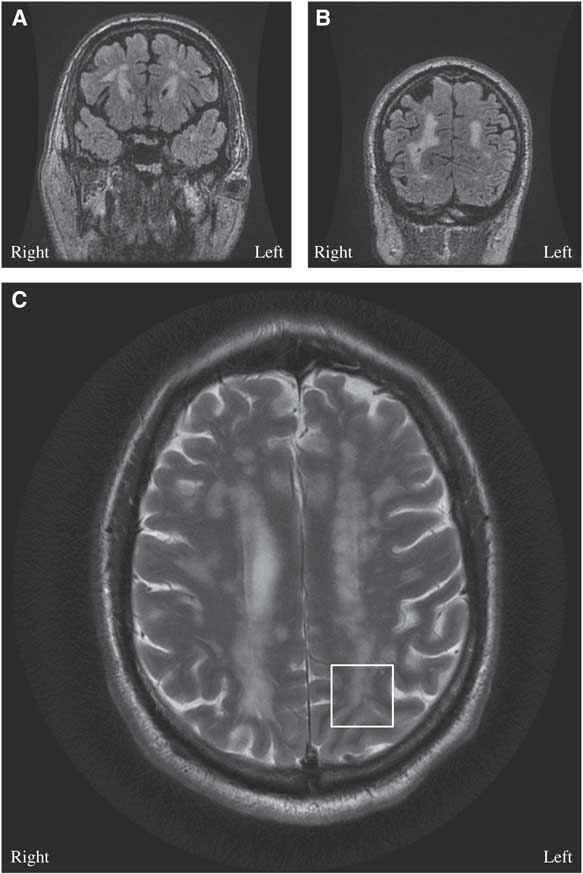
Figure 1 (A, B) Brain MRI coronal T2 Fluid Attenuation Inversion Recovery (FLAIR)-multifocal confluent and discrete subcortical, deep and periventricular lesions are seen involving both cerebral hemispheres, including temporal and occipital lobes. (C) Brain MRI axial T2: few lesions are seen extending through the U fibers and abutting the cortex, notable in the left occipital cortex (white box).
The diagnosis of cerebral autosomal dominant arteriopathy with subacute infarcts and leukoencephalopathy (CADASIL) was entertained on the basis of the history of migraine with aura, family history of early-onset ischemic strokes and diffuse white matter disease on MRI. On genetic analysis, he was found to be heterozygous for a NOTCH3 mutation, C619C>T, and it is pathogenic for CADASIL.Reference Oberstein, Ferrari and Bakker 1 His atypical visual auras were consistent with focal epilepsy originating from the juxtacortical lesions in the left occipital lobe (Figure 1C).
CADASIL is an autosomal dominant inherited angiopathy due to mutations in the NOTCH3 gene on chromosome 19. Migraine with aura is often the initial manifestation of CADASIL, and auras can present with atypical features including acute-onset, long-lasting, isolated motor, sensory, visual or brainstem auras.Reference Chabriat, Joutel, Dichgans, Tournier-Lasserve and Bousser 2
Epilepsy is rare in CADASIL, and it has only been reported in 10% of patients.Reference Dichgans, Mayer and Uttner 3 , Reference Haan, Oberstein and Ferrari 4 The postulated pathophysiology is cortical or juxtacortical involvement—that is, subcortical lacunar infarcts secondary to angiopathy at the gray-white matter junction (Figure 1C).Reference Dichgans, Mayer and Uttner 3 - Reference Bentes, Pimentel and Ferro 5 In a case series with 102 patients with CADASIL, 10 patients had epileptic seizures and nine of them had a history of strokes.Reference Dichgans, Mayer and Uttner 3
Our patient was transitioned to oral levetiracetam at a dose of 500 mg twice daily with no further seizures. He was referred for genetic counseling and his follow-up EEG in 2 months was normal. Lifestyle modifications to decrease stroke risk factors were re-emphasized. Although epilepsy is rare in CADASIL, CADASIL should be on the differential diagnosis in the context of history of migraine with atypical aura, ischemic strokes, family history of early-onset ischemic strokes, diffuse white matter disease and particularly juxtacortical ischemic lesions on MRI.
DISCLOSURES
TLHC and MS report no disclosures and have no conflicts of interest. JGB has received financial support for research and educational activities from UCB Canada and Eisai, outside the submitted work.
STATEMENT OF AUTHORSHIP
TLHC examined the patient. All authors wrote and revised the manuscript and created the figures together.

