



The neuronal ceroid-lipofuscinoses (NCLs) are a heterogeneous group of inherited neurodegenerative disorders in which abnormal lipopigments are deposited to form lysosomal inclusion bodies in neurons.Reference Bennett and Rakheja 1 The major clinical findings in affected patients include seizures, visual loss from retinal degeneration, regression of intellectual and motor skills, and premature death.Reference Nardocci, Verga, Binelli, Zorzi, Angelini and Bugiani 2 Most NCLs are inherited in an autosomal recessive fashion and are present in infancy or childhood. However, there is a rare adult-onset form referred to as Kufs disease, which differs from the pediatric-onset NCLs by the absence of visual loss. Typical symptoms include seizures, psychiatric manifestations, and progressive dementia,Reference Velinov, Dolzhanskaya and Gonzalez 3 often presenting in the third or fourth decade of life.Reference Berkovic, Carpenter, Andermann, Andermann and Wolfe 4 Although Kufs disease is frequently inherited in an autosomal recessive fashion,Reference Arsov, Smith and Damiano 5 autosomal dominant Kufs disease (ADKD) has also been reported.Reference Velinov, Dolzhanskaya and Gonzalez 3
A small number of affected families with ADKD have been described in the United States, Spain, Netherlands, and Czech Republic.Reference Benitez, Alvarado and Cai 6 - Reference Sims, Cole, Sherman, Caruso and Snuderl 11 The affected family members presented with generalized seizures, movement disorders, and dementia. The age of onset varied from 25 to 46 years. Mutations in the DNAJC5 gene have recently been reported to be a cause of ADKDReference Burneo, Arnold, Palmer, Kuzniecky, Oh and Faught 7 , Reference Boehme, Cottrell, Leonberg and Zeman 12 - Reference Josephson, Schmidt, Millsap, McManus and Morris 14 in about 25% of cases.Reference Arsov, Smith and Damiano 5
This gene codes for the highly conserved cysteine string protein α (CSPα), so named for a cysteine-rich region (14 of 24 amino acids) that is palmitoylated through thioester linkages with cysteine side chains. This arrangement allows CSPα to attach to membranes and function as a chaperone protein that controls the vesicular exocytosis machinery, particularly at the synapse.Reference Chamberlain and Burgoyne 15 Here, we present a family affected with ADKD resulting from a previously reported DNAJC5 gene mutation.Reference Burneo, Arnold, Palmer, Kuzniecky, Oh and Faught 7 , Reference Boehme, Cottrell, Leonberg and Zeman 12 - Reference Josephson, Schmidt, Millsap, McManus and Morris 14 We describe the clinical profiles of several members in this family, the pedigree, and the neuropathology in one of the members (the proband) who came to autopsy. Because the cholinergic system is involved in a number of functions, including cognition, we also examined the cholinergic system in the brain of the proband.
Materials and Methods
Case Histories
In this study, we report on one family with ADKD (Figure 1). Following approval from the Research Ethics Boards of Horizon Health Network and Western University in London, Ontario, patients or their substitute decision-makers provided informed consent to have their charts and results of their genetic testing reviewed. The genetic testing undertaken, as part of usual care, consisted of sequencing of the DNAJC5/CLN4 gene.
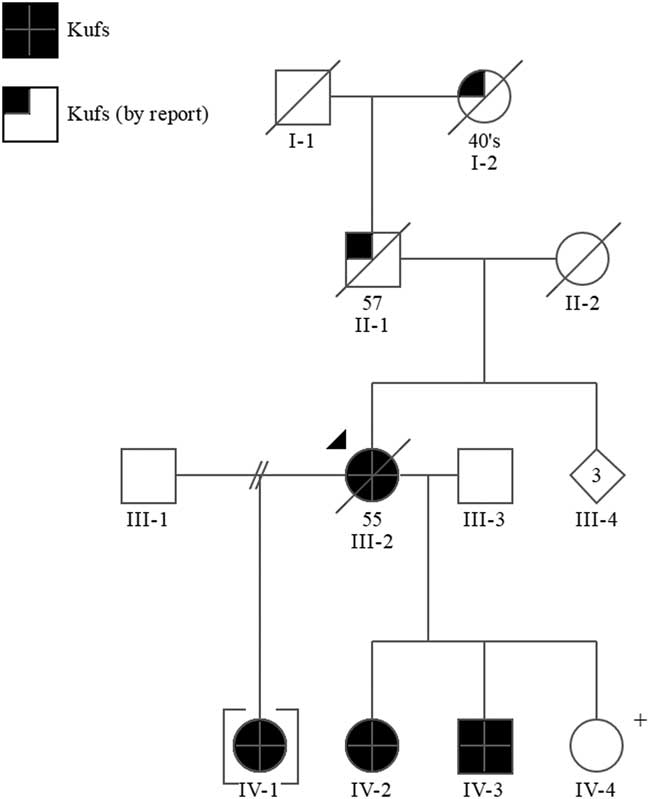
Figure 1 Pedigree. +=positive predictive genetic test.
III-2 (Proband)
This woman had been well until age 36 years, when she developed seizures that began with colored lights and then evolved into a generalized seizure; she was placed on anticonvulsants. At age 42, she had a “big seizure” that was followed by both physical and cognitive impairment. She developed marked decline in gait, which was described as shuffling, with reduced arm swing. Her cognitive decline began with repetition of questions and forgetting conversations. Her functional abilities declined, so that she required moderate assistance with activities of daily living (ADLs) and significant assistance with instrumental activities of daily living (IADLs). She also developed a stutter and auditory hallucinations. Over the course of several years, she was admitted three times to a psychiatric unit and prescribed olanzapine for hallucinations. She also developed myoclonic jerks and tremors. A magnetic resonance imaging (MRI) scan of the brain showed moderate to severe global cerebral and cerebellar atrophy, which was out of keeping with the patient’s age.
At age 43, she had diffuse cognitive impairment with difficulties in attention and concentration, orientation, naming, writing, visual spatial function, and frontal system tasks. By age 46, her seizures were under reasonable control but her functional abilities deteriorated to the point that she required assistance with personal care and was incontinent. At age 53, she was living in a nursing home and required total care for all basic needs. She had significant myoclonus and was wheelchair bound. Although she had speech difficulties with stuttering, she could still communicate with difficulty; for example, she could still sing the lyrics to her favorite country music singers when she listened to the song and she could still name her children. Her seizures were controlled with phenytoin and valproic acid; she was also on benztropine, olanzapine, and venlafaxine. The etiology for her presumed neurodegenerative disorder at that time was not clear. Molecular genetic testing for the presenilin-1 (PSEN1) gene for Alzheimer disease and the prion protein gene (PRNP) for familial Creutzfeldt-Jakob disease was negative. She continued to decline in the nursing home and she died there at age 55. Autopsy showed neuronal storage disease, consistent with Kufs disease.
The brain of III-2 was examined at autopsy; it was removed 37.5 hours after death and fixed in formalin for 317 days. According to the family’s wishes on her behalf, half of the brain was donated to the Maritime Brain Tissue Bank for research. As part of continued research on neurochemical and neuroanatomical changes in neurodegenerative disorders, this half of the brain was examined for changes in neurotransmitter systems and neuroanatomical changes using immunohistochemical methods. Studies followed approval by the Nova Scotia Health Authority Research Ethics Board.
IV-2
This woman is the proband’s second-born child and is a half sibling to IV-1 and full sibling to IV-3 and IV-4. She had been well until the age of 31 years, when she began to have difficulty at work forgetting codes and developed “clumsiness”; she was obliged to stop work. She also developed “twitching” during sleep. Two years later, she presented with a possible seizure and an electroencephalograph (EEG) revealed generalized epileptiform discharges that appeared to be activated by hyperventilation. She was started on valproic acid with good control of her seizures.
At age 33, she began to have difficulties with word finding and right-sided headaches. At that time, she scored 23 of 30 on the Mini-Mental State Examination (MMSE). Her difficulties were spelling “world” backwards, repeating “no ifs, ands or buts,” and the intersecting pentagon. Her clock drawing was also impaired. She scored 17 of 30 on the Montreal Cognitive Assessment (MoCA), having difficulties with cube drawing, naming, word recall (1 of 5), repeating sentences, subtraction, and digit span. Funduscopic examination was reported as normal with no Kayser-Fleischer ring externally or with gonioscopy.
Functionally, she was completely independent for her ADLs, and could walk independently, but required assistance with all her IADLs. An MRI scan of her brain showed moderate global cerebral and cerebellar atrophy as well as small foci of globus pallidus calcification. She had postural and action tremors of her right upper limb. She underwent skin and muscle biopsies, which were normal. Neuropsychological testing showed impairment in general intelligence, short-term memory, receptive and expressive language skills, attention, visual-spatial skills, and executive functioning. She was treated for depression with citalopram, which improved her mood. Over the next year, she developed more difficulty with slurring of her speech and her myoclonus continued, particularly at night. Her seizures remained under control, but she had worsening of her gait and balance.
Over the next 2 years, she continued to show a decline in her language skills and mobility; she started using a walker at age 36. She continued to show deterioration in her gait and speech until, at age 38, she required assistance with her personal care and was only able to walk short distances with a walker. Her MMSE at this time was 21/30. At age 39, she continued to need assistance with her ADLs and was dependent for her IADLs. An EEG at that time showed symmetrical alpha rhythm of eight to nine cycles per second dominant over the posterior head regions. General background activity showed an increase in theta and occasional delta activity, with occasional sharp waves over the temporal region bilaterally. Currently, her epileptic seizures remain under good control but her myoclonic jerks persist and can occur at any time of the day and often result in a fall.
IV-3
This man is the only son to the proband III-2 and had a history of alcohol misuse until age 30 years when he was admitted to hospital after suffering his first probable epileptic seizure. He remembered seeing sparkling lights and feeling odd just before losing his awareness. Cranial computed topography and MRI scans at this time were normal; however, his EEG demonstrated multiple brief bursts of fragments of generalized epileptiform discharges. He was started on phenytoin. At age 32, he scored 23 of 30 on the MMSE, losing points in orientation, attention, language, visual spatial, and clock drawing. He scored 14/30 on the MoCA, losing points on the visual spatial, executive functioning, naming, attention, language, abstraction, and delayed recall tasks. His speech was noted to be slightly dysarthric, noticeably having trouble forming his Rs properly, but he did not have a stutter. He did not require a walking aid, his balance was within normal limits, and he was independent for his ADLs and IADLs. He completed a modified grade 12 education program. He was not diagnosed with a specific learning disability as a child, but was reported to have had trouble reading and writing throughout school. He continued to have generalized tonic clonic seizures; however, he was not compliant with his medications and had only recently stopped drinking alcohol. At age 33, he scored only 8 of 30 on the MoCA; however, it was noted that he had poor effort throughout testing and he frequently expressed statements that he could not complete the tasks. He had ongoing myoclonus that was becoming more troublesome and would wake him up from sleep. An EEG showed generalized sharp activity throughout all the leads, with a slight left temporal predominance. At age 35, he was continuing to have seizures, and now lives in a boarding house in the community where he receives assistance with his meals and medications. He is able to do his own ADLs but is dependent for all of his IADLs.
IV-1
This woman is the firstborn child to the proband III-2 and a half-sibling to IV-2, IV-3, and IV-4, and was given up for adoption at birth. She was healthy until the age of 33 years when she began to experience myoclonic jerks that occasionally resulted in falls. The falls progressively increased in frequency so that they occurred every time she tried to walk and myoclonic jerks became almost constant.
At age 38, she developed generalized tonic clonic seizures. At that time, cranial MRI scans revealed mild generalized brain atrophy; EEG revealed generalized spikes and waves. A month later, detailed neuropsychological evaluation showed widespread cognitive impairment across most tasks, but normal range visual memory. She scored 14 of 30 on the MoCA, where she had significant deficits in visual-spatial tasks, executive functioning, delayed recall, attention, serial seven subtractions, and naming. She was unable to walk and became confined to a wheelchair.
At age 39 years, the patient underwent video-EEG telemetry recordings for 5 days. These recordings showed abundant generalized spikes and waves independent of her myoclonic jerks and normal sleep architecture. Repeat cranial MRI scans revealed moderate diffuse brain atrophy.
IV-4
The youngest child of III-2 was clinically asymptomatic at the age of 28. After genetic counseling, she underwent predictive genetic testing and was found to be positive for the familial mutation in the DNAJC5 gene. An MRI scan of her head showed decreased signal intensity in the region of the putamen and an EEG showed moderately severe slowing in keeping with severe diffuse disturbance in cerebral function.
Additional Family History
The proband’s father (II-1) died at the age of 57 years in a psychiatric institution with what was thought to be Alzheimer disease. He had a history of epileptic seizures and was wheelchair bound. Her mother (II-2) was thought to have had schizophrenia and possible Parkinson’s disease, but the immediate cause of her death was unknown. The proband’s paternal grandmother (I-2) was reported to have a history of “fits” and died between the ages of 45 and 50.
Genetic Testing
Sequence analysis of the DNAJC5/CLN4 gene in the proband III-2 identified a mutation, c.346_348delCTC(p.L116del), which has been reported in other individuals with Kufs disease.Reference Benitez, Alvarado and Cai 6 Subsequent targeted mutation analysis in IV-1, IV-2, IV-3, and IV-4 confirmed they are heterozygous for this mutation.
Neuropathological Examination
The brain of III-2 (proband) was removed at autopsy and immerse fixed in 10% formalin for 317 days before sectioning. It weighed 1000 g. Tissue sections were paraffin embedded and examined initially by staining for hematoxylin and eosin/Luxol fast blue. Additional stains (for periodic acid–Schiff, Sudan Black, Bielschowsky silver stain, and immunostaining for tau, and pathogenic prion protein) were performed.
For comparison, tissue was obtained from the brain of a 66-year-old female who had no clinical history of any neurological condition. The brain was fixed for 330 days. The brain was obtained from the Maritime Brain Tissue Bank as part of continued research on neurochemical and neuroanatomical changes in neurodegenerative disorders, after approval by the Nova Scotia Health Authority Research Ethics Board. This brain weighed 1325 g and was normal upon neuropathological examination.
Immunohistochemistry for Acetylcholinesterase and Choline Acetyltransferase
Acetylcholinesterase (AChE) was visualized in the brain sections from both proband and normal control using immunohistochemistry. Sections were rinsed in 0.1 M phosphate buffer (PB), pH 7.4 for 30 minutes, placed in 0.3% H2O2 in PB for 30 minutes to quench endogenous peroxidase activity, and rinsed again for 30 minutes. Sections were incubated for 1 hour in PB with 0.1% Triton X-100 and normal goat serum (NGS, 1:100). Sections were incubated overnight at room temperature in PB with 0.1% Triton X-100, NGS (1:100; Sigma), and mouse anti-AChE primary antibody (1:500; Millipore, MAB303) for about 17 hours. After rinsing, sections were incubated in PB with 0.1% Triton X-100, NGS (1:1000), and biotinylated goat anti-mouse secondary antibody (1:500; Vector Labs) for 1 hour. After another rinse, sections were placed in PB with 0.1% Triton X-100 and Vectastain Elite ABC kit (Vector Labs) according to the manufacturer’s instructions. After rinsing, sections were developed in a PB solution containing 1.39 mM 3,3′-Diaminobenzidine tetrahydrochloride (DAB). After 5 minutes, 50 μl 0.3% H2O2 in dH2O was added per milliliter of DAB staining solution and was incubated for 2 to 5 minutes. The reaction was stopped by rinsing the sections in 0.01 M acetate buffer, pH 3.3. Sections were mounted on slides, cleared in xylene, and cover-slipped. Control experiments were performed by omitting the primary antibody.
Choline acetyltransferase (ChAT) was also visualized using immunohistochemistry. Free-floating sections were rinsed in 0.05 M Tris buffered saline (TBS), pH 7.6, for 30 minutes, placed in 0.3% H2O2 in TBS for 30 minutes to quench endogenous peroxidase activity, and rinsed again for 30 minutes. For antigen retrieval, sections were heated to 80°C for 30 minutes in 10 mM citrate buffer pH 6.0 with 0.05% Tween20 (Sigma) and allowed to cool to room temperature. Sections were rinsed in TBS for 30 minutes and then incubated for 1 hour in TBS with 0.1% Triton X-100 and normal rabbit serum (NRS, 1:25). Sections were incubated overnight at room temperature in TBS with 0.1% Triton X-100, NRS (1:100; Sigma) and goat anti-ChAT primary antibody (1:1000; Millipore, AB144P) for about 17 hours. After rinsing, sections were incubated in TBS with 0.1% Triton X-100, NRS (1:1000), and biotinylated rabbit anti-goat secondary antibody (1:500; Vector Labs) for 1 hour. After another rinse, sections were placed in TBS with 0.1% Triton X-100 and Vectastain Elite ABC kit (Vector Labs) according to the manufacturer’s instructions. After rinsing, sections were developed in a solution of TBS (pH 8.0) containing 1.39 mM DAB and 0.6% nickel ammonium sulfate. After 5 minutes, 50 μl 0.3% H2O2 in dH2O was added per milliliter of DAB staining solution and was incubated for 3 to 5 minutes. The reaction was stopped by rinsing the sections in TBS. Sections were mounted on slides, cleared in xylene, and cover-slipped. Control experiments were performed by omitting the primary antibody.
Data Analysis
The stained sections were analyzed and photographed using a Zeiss Axioplan 2 motorized microscope with a Zeiss Axiocam HRc digital camera using AxioVision 4.6 software (Carl Zeiss Canada Ltd., Toronto, Ontario, Canada). The photographs were assembled using Adobe Photoshop. The images were color balanced and contrast enhanced, and the brightness was adjusted to match the background from different images.
Ethical Approval
All procedures performed involving human participants were in accordance with the ethical standards of the institutional research committees and with the 1964 Helsinki declaration and its later amendments. Informed consent was obtained from all individual participants included in the study.
Results
Neuropathology
The unfixed ADKD brain weighed 1000 g, which is considerably less than an average, normal brain. Gross examination of the formalin-fixed brain showed diffuse atrophy of the frontal and temporal lobes and a lesser degree of atrophy of the parietal and occipital lobes. On coronal sectioning of the cerebral hemispheres, there was pallor of the subcortical white matter and sparing of the subcortical U fibers. Atrophy resulted in marked rounding of the frontal horns of the lateral ventricles. Horizontal sections through the brainstem showed pallor of the substantia nigra relative to the locus ceruleus. Paramedian sections through the cerebellum showed generalized hemispheric atrophy. Microscopic examination showed a striking increase, relative to control, in neuronal storage of golden brown material resembling lipofuscin (Figure 2A). The neuronal storage material was stained strongly by periodic acid–Schiff and Sudan black stains, characteristic of lipofuscin (Figure 2B and 2C). Importantly, there were no amyloid plaques or neurofibrillary tangles (ruling out Alzheimer disease) and cortical immunostaining for pathogenic prion protein was negative (ruling out a prion disease). Ultrastructural examination (by scanning electron microscopy) showed granules of electron-dense and electron-lucent material in cortical neurons, consistent with lipofuscin (Figure 2D). The final neuropathological diagnosis was neuronal storage disorder consistent with Kufs disease.
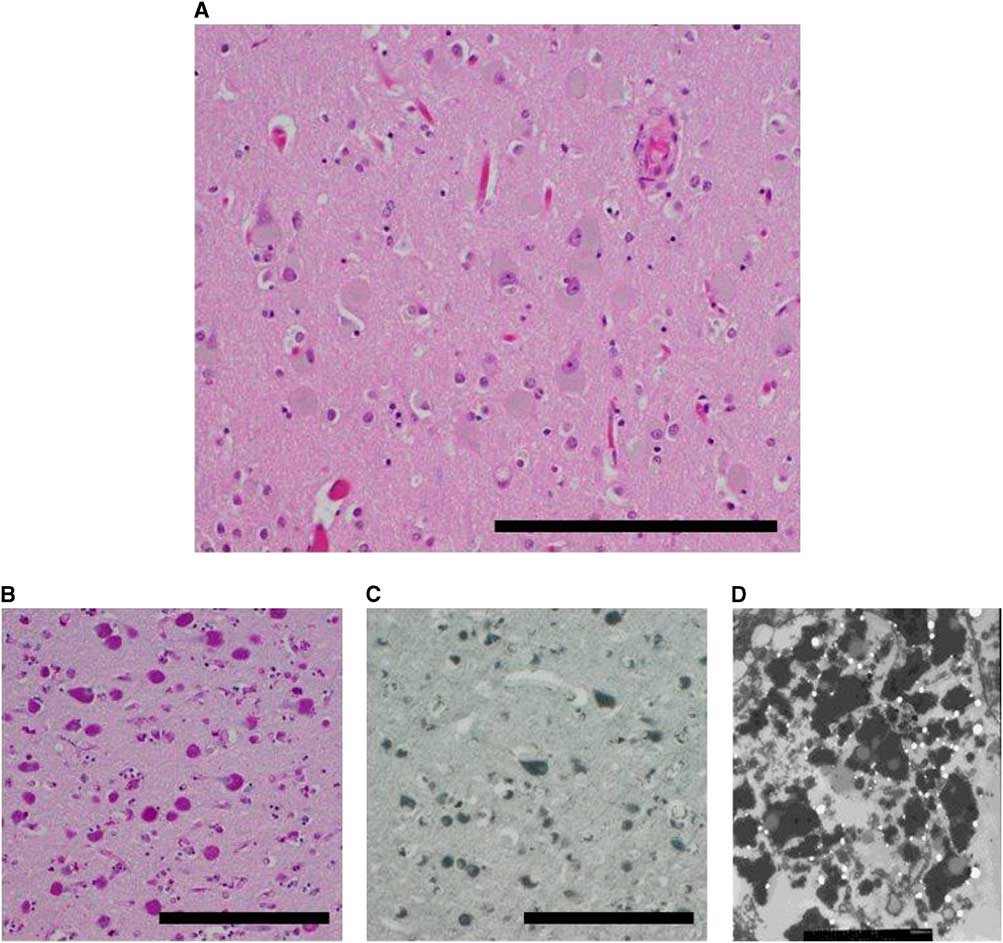
Figure 2 Neuropathology. (A) Cortical neurons distended by brown material (right parietal lobe). (B) Material is positive for periodic acid–Schiff stain (PAS). (C) Material is also positive for Sudan Black B stain. (D) Electron micrograph reveals lipofuscin-like material. Scale bars, 100 µm.
AChE
The basal ganglia (caudate nucleus, putamen, and the internal and external segments of the globus pallidus) in the normal brain was richly stained for the enzyme AChE (Figure 3A), as has been described previously.Reference Graybiel and Ragsdale 16 In III-2 (proband), there was profound loss of this enzyme in all components of the basal ganglia (Figure 3B). The normal basal forebrain, particularly the nucleus basalis of Meynert, was heavily stained for AChE (Figure 3A), as has been described previously.Reference Mesulam and Geula 17 For III-2, the basal forebrain exhibited AChE staining that was comparable to control (Figure 3B).
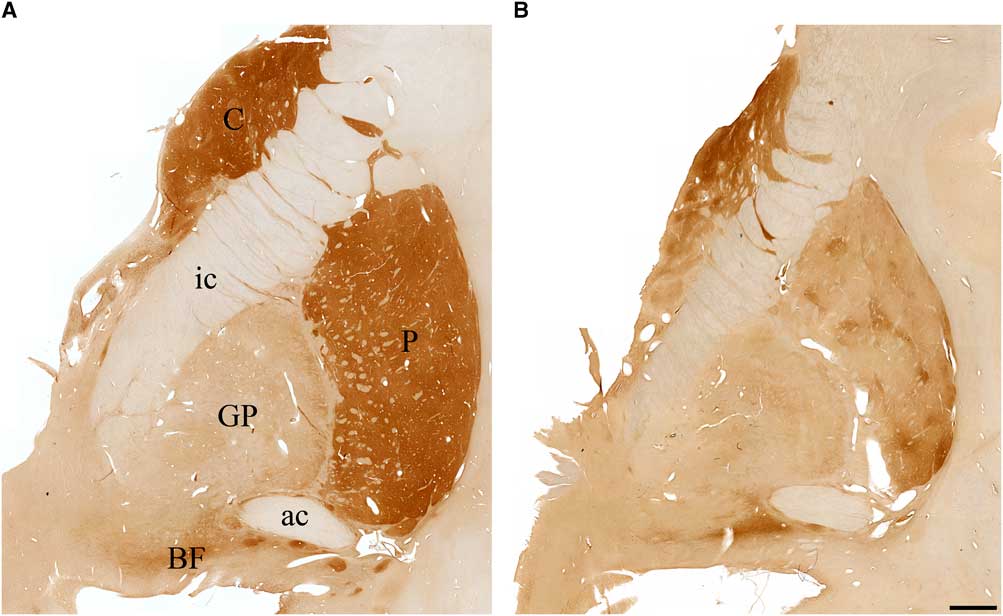
Figure 3 Photomicrographs of coronal sections stained for acetylcholinesterase in (A) a cognitively normal 66-year-old female and (B) the proband with Kufs disease. Note significant loss of acetylcholinesterase staining in caudate (C), putamen (P), globus pallidus (GP), and the basal forebrain (BF) in the proband brain compared with control. ac=anterior commissure; ic=internal capsule.). Scale bar, 2.5 mm.
Choline Acetyltransferase
Normal basal ganglia had cholinergic interneurons and neuropil stained for ChAT (Figure 4A), as has been shown previously.Reference Holt, Graybiel and Saper 18 There was no difference observed in neuronal staining in the basal ganglia between normal and Kufs disease but there was some reduction in neuropil staining (Figure 4A and 4B). In the normal Nucleus Basalis of Meynert there was robust staining for ChAT (Figure 4C). In contrast, in Kufs disease, there was a profound loss of staining for this enzyme (Figure 4D).
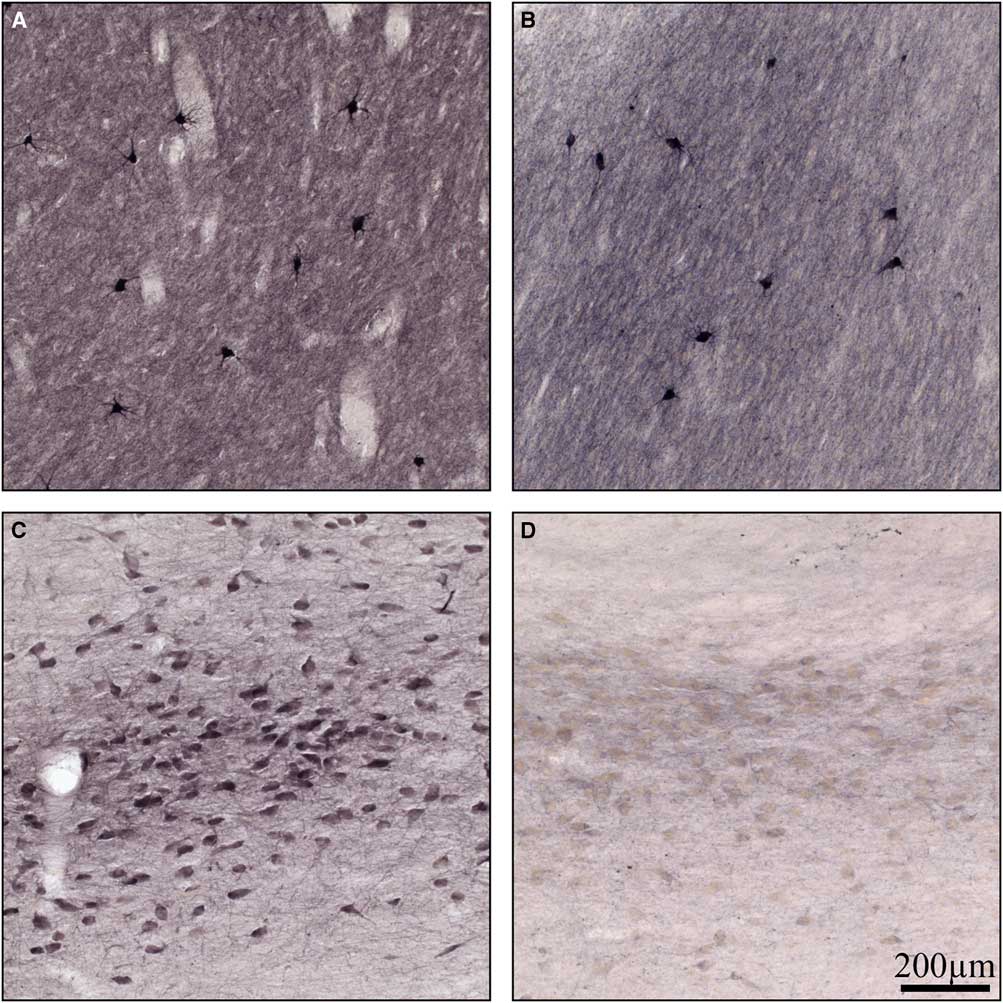
Figure 4 High-power photomicrographs of coronal sections stained for choline acetyltransferase in (A) normal basal ganglia, (B) Kufs basal ganglia, (C) normal basal forebrain, and (D) Kufs basal forebrain. Note no loss of choline acetyltransferase staining in Kufs basal ganglia, but profound loss in Kufs basal forebrain. Scale bar, 200 µm.
Discussion
We report a family in which several members developed cognitive impairment, seizures, hallucinations, myoclonus, and gait impairment. The diagnosis of NCL was realized in the proband at autopsy (Figure 2). Subsequent genetic testing revealed that all affected individuals had a mutation in the DNAJC5 gene. The DNAJC5 gene encodes CSPα, which is a member of the DNAJ/HSP40 family of cochaperone molecules that are essential for transport of vesicles at the presynaptic terminal.Reference Benitez, Alvarado and Cai 6 CSPα is a highly conserved protein that interacts with many other cochaperones, including HSP70Reference Nosková, Stránecký and Hartmannová 10 and those required for exocytosis of vesicles in neurotransmission.Reference Fernández-Chacón, Wölfel and Nishimune 19 CSPα is a 198 amino acid residue protein that has a phosphorylation domain (at serine 10), a J domain for binding HSC70, and cysteine-string domain for amino acids residues 112-137. These cysteine amino acid residues become palmitoylated for membrane binding. It has been shownReference Tiwari, d’Orange and Troakes 20 that CSPα facilitates formation of the SNARE complex, part of the machinery necessary for exocytosis of vesicles.
It is now establishedReference Burgoyne and Morgan 21 that CSPα is essential for prevention of neurodegeneration. For example, CSPα knockout mice appear normal at birth (and remain normal for a few weeks) but then develop progressive sensory motor disorders and die within 8 weeks.Reference Fernández-Chacón, Wölfel and Nishimune 19 These mice show extensive rapid degeneration of retinal photoreceptorsReference Schmitz, Tabares and Khimich 22 and hippocampal GABAergic neurons.Reference García-Junco-Clemente, Cantero and Gómez-Sánchez 23 Another important observation in these knockout mice is that the lethal neurodegeneration caused by the deletion of CSPα is prevented by the transgenic expression of α-synuclein. It is interesting that this CSPα knockout phenotype is exacerbated when α-synuclein gene is also knocked out.Reference Chandra, Gallardo, Fernández-Chacón, Schlüter and Südhof 24 It appears that α-synuclein is another cochaperone molecule that is essential for transport of vesicles to the presynaptic membrane and can compensate for lack of CSPα. In other neurodegenerative conditions, CSPα has been shownReference Miller, Swayne and Chen 25 , Reference Shirasaki, Greiner and Al-Ramahi 26 to interact with Huntingtin protein, but its relevance in Huntington disease is yet to be established. It has also been reportedReference Sims, Cole, Sherman, Caruso and Snuderl 11 that expression of CSPα is reduced in the Alzheimer disease forebrain in postmortem analysis. In adult-onset neuronal ceroid lipofuscinosis, with mutations in the gene encoding CSPα, DNAJC5, it has been shown by a number of groups to be a gene causing ADKD.Reference Velinov, Dolzhanskaya and Gonzalez 3 , Reference Benitez, Alvarado and Cai 6 , Reference Cadieux‐Dion, Andermann and Lachance‐Touchette 8 , Reference Nosková, Stránecký and Hartmannová 10 This condition, also referred to as Parry’s disease, is a rare neurodegenerative disorder; therefore, this work adds to the slowly growing body of literature about this particular condition, how it can be diagnosed, and its consequences at the molecular and neuroanatomical levels.
The mutation in the family described here leads to deletion of amino acid residue 116 within the cysteine string domain CSPα. Presumably, this leads to inability for CSPα to efficiently bind to vesicles for their transport to the presynaptic membrane. At present, it is not known with certainty which neuronal types degenerate in this CSPα mutation in humans.
Given that there is significant cognitive impairment in Kufs disease and that cognition is highly dependent on an intact cholinergic system,Reference Everitt and Robbins 27 we examined the brain of one individual and the distribution of the cholinergic markers, ChAT and AChE, compared with normal control brain. Whether other neurotransmitter systems are also affected will require further examination of this Kufs disease brain in the future. Clearly, in this condition, the cholinergic interneurons and cholinergic projection fibers within the basal ganglia are spared (Figure 3), but there is profound loss of ChAT within the basal forebrain (Figure 4). Within the basal forebrain, based on the presence of lipofuscin-laden neuronal profiles, there appears to be loss of ChAT rather than loss of neurons themselves. Using AChE immunohistochemistry, there is also profound loss of this enzyme within caudate, putamen, and basal forebrain regions. Cholinergic hypofunction typically does not lead to seizures or myoclonus. In fact, it is increased cholinergic tone that contributes to these phenomena. It is therefore unlikely that the deficit in cholinergic system observed here is a pathophysiological factor in causing seizures and myoclonus in Kufs disease. Other degenerative processes, including synaptic loss, may be involved in these phenomena. This loss of cholinergic markers suggests that, at least in part, cognitive impairment in these individuals with Kufs disease is a consequence of impairment of cholinergic neurotransmission in the brain.
Conclusion
The postmortem brain in Kufs disease (ADKD/Parry’s disease), with a pathogenic L116 deletion mutation in the DNAJC5/CLN4 gene, demonstrates profound loss of cholinergic neurotransmission that may account, in part, for a number of signs and symptoms observed in this condition. Because of the rare nature of Kufs disease, only one brain was available for examination and, as a preliminary study, we chose to compare the cholinergic system of the Kufs brain to a normal control under comparable conditions. How changes in CSPα function leads to a loss of cholinergic neurotransmission is as yet unknown, but the present work, showing loss of enzymes involved in acetylcholine metabolism, provides a basis for further examination of the role of CSPα in neurotransmission.
Acknowledgments
We thank Andrew Reid for his excellent technical assistance and the Maritime Brain Tissue Bank (CORES Facilities, Faculty of Medicine, Dalhousie University) for brain tissues. In addition, we thank the family members who graciously agreed to take part in this research.
Disclosures
The authors declare that they have no conflict of interest.




