



Introduction
Spinal dysraphism is a broad term that encompasses a number of conditions affecting the spine and spinal cord. Combined, these disorders are the second most common congenital malformations, after heart defects. Reference Copp and Greene1,Reference Au, Ashley-Koch and Northrup2 Most dysraphic conditions are associated with abnormalities of neural tube formation and are grouped together as neural tube defects (NTDs). These include myelomeningocele (MMC), dermal sinus tracts, lipomyelomeningocele, spinal cord lipoma, split cord malformation (SCM), and others. Reference Acharya, Pendharkar, Varma, Pruthi and Varadarajan3 A meningocele is an example of a dysraphic condition that affects the meninges and dorsal vertebral elements but not the spinal cord and is not usually defined as a NTD. The clinical manifestations of spinal dysraphism vary greatly and patients can range from asymptomatic to severely disabled. The embryologic origin of the anomaly is often directly related to the severity of disability and also impacts the appropriate surgical treatment.
Spinal dysraphism has a substantial societal impact. Previous work from Ouyang et al. estimated the lifetime cost for medical expenditures among patients with MMC as greater than $300 000 (USD). Reference Ouyang, Grosse, Armour and Waitzman4 While the initiation of folic acid fortification has successfully decreased the incidence of NTDs, it has not prevented them. Reference Grosse, Berry, Mick Tilford, Kucik and Waitzman5 Importantly, the anatomic abnormality encountered in the patient is directly related to a defect that occurred during embryogenesis. A clear understanding of the dysraphic condition’s origin is a necessary requirement for appropriate treatment selection and providing the patient and family with a range of anticipated functional outcomes. This review will describe fundamental embryology of the central nervous system (CNS) and then provide a more detailed discussion of how the embryologic anomaly affects clinical decision-making.
Neurulation
Classic Neuroembryology
The end of gastrulation marks the beginning of development of the CNS and takes place at the start of the third week of gestation. Reference Schoenwolf, Bleyl, Brauer, Francis-West, Schoenwolf, Bleyl, Brauer and Francis-West6,Reference Carlson and Carlson7 At this stage, the embryo is a flat bilayered disk. The primitive streak develops on the epiblast and proceeds in a caudo–cranial direction. This process establishes the body plan of the embryo related to the cranial and caudal direction and the left-to-right axis. As the streak progresses, cells at its anterior limit begin to coalesce and form the primitive node (Hensen’s node). Reference Viebahn8 At the same time, epiblast cells migrate near the primitive streak and pass through it, leading to formation of the three distinct germ layers. The streak eventually ingresses and leads to the formation of the notochord which in turn induces the formation of the neural crest and neural tube. Reference Carlson and Carlson7,Reference Wolpert, Tickle and Arias9 This process is complex and believed to be influenced by over 200 identified genes. Reference Copp, Greene and Murdoch10,Reference Juriloff and Harris11 The formation of the neural tube, which forms the majority of the CNS, is defined as neurulation. Reference Greene and Copp12,Reference Schoenwolf and Smith13 After the neural tube has closed, it separates from the overlying cutaneous ectoderm by a process called dysjunction. Reference Kaufman14,Reference Selcuki, Vatansever, Umur, Temiz and Sayin15 While the cranial portion of the spinal cord forms from primary neurulation, the caudal spinal cord and conus medullaris arise from secondary neurulation. This process begins around day 28 with canalization of the caudal cell mass (CCM), a collection of mesenchymal cells caudal to the neural tube. As the CCM canalizes, it eventually becomes continuous with the neural tube and leads to the aforementioned neural elements. Reference Schoenwolf16–Reference Muller and O’Rahilly18
Convergent Extension
Although open NTDs result from failure of primary neurulation, there are a number of steps that precede neural tube closure and allow for that development to take place. While the embryo is still a bilaminar disk, it becomes elongated and flattened via a process called convergent extension. Reference Keller19,Reference Keller, Shook and Skoglund20 This shaping of the disk is essential for proper neural tube closure, and multiple animal models have demonstrated that failure of convergent extension leads to craniorachischisis, in which almost the entire neuroaxis (from midbrain to low spine) is exposed. Reference Copp, Greene and Murdoch10,Reference Ybot-Gonzalez, Savery and Gerrelli21,Reference Lei, Zhang, Li, Wu, Jin and Wang22 In addition to this severe NTD, defective convergent extension leads to a truncated rostro-caudal length as the disk fails to properly elongate. Reference Copp and Greene1
The planar cell polarity pathway is a key molecular regulator of convergent extension and relies on noncanonical Wnt signaling cascades, in which frizzled membrane receptor activation leads to phosphorylation of the disheveled (DVL) cytoplasmic signaling molecule. Reference Copp and Greene1,Reference Montcouquiol, Crenshaw and Kelley23,Reference Ciruna, Jenny, Lee, Mlodzik and Schier24 Studies in both Xenopus species and mice have demonstrated the importance of these pathways in neurulation. Impaired convergent extension via inhibition of the planar cell polarity pathway has been associated with craniorachischisis and other open NTDs in both animal models and humans. Reference Juriloff and Harris11,Reference Wallingford and Harland25 For a more comprehensive overview of the genetics related to convergent extension and its relationship to neurulation, readers are directed to the review by Copp and Greene. Reference Copp and Greene26
Neural Plate Bending
After the neuroepithelium has elongated, it bends at specific points, allowing the prominences of the neural folds to come closer together and eventually close. Reference Greene and Copp27 However, mouse models have demonstrated different patterns of bending based on the rostral-caudal location of the developing embryo. The midbrain bends at both a medial hinge point (MHP) and a dorsolateral hinge point (DLHP); the upper spine at only a MHP; the mid-spine at both a MHP and a DLHP, and the lower spine only at a DLHP. Reference Shum and Copp28
Cells of the MHP and DLHP grow and divide at different rates, creating the neural folds that can allow for the neural tube to close properly. These processes are regulated by a complex interplay between the underlying notochord and overlying ectoderm, as well as signaling molecules Sonic hedgehog (Shh), bone morphogenic protein-2 (BMP2), and noggin (Figure 1). Reference Ybot-Gonzalez, Cogram, Gerrelli and Copp29–Reference Eom, Amarnath, Fogel and Agarwala33 Experimental evidence suggests that Shh and BMP prevent formation of the DHLP, and that overexpression in the lower spine leads to MMC. Reference Stottmann, Berrong, Matta, Choi and Klingensmith34 Lastly, depending on the cranial–caudal location of the developing embryo, changes in cellular architecture also contribute to neural fold bending. Reference Greene and Copp27,Reference Morriss-Kay and Tuckett35 Actin monofilaments, primarily under the influence of Shroom, play a particularly important role in the closure of the cranial neuropore. Reference Ybot-Gonzalez and Copp36,Reference Haigo, Hildebrand, Harland and Wallingford37

Figure 1: Representation of the different molecular substances affecting the MHP and DLHP. Adapted with permission from refs. 26 and 30.
Neural Tube Fusion
Once the neural folds have met in the midline and are opposed, they must fuse in order to complete the closure of the neural tube. Reference Pai, Abdullah and Mohd-Zin38 Waterman demonstrated that the adjacent epithelial cells change shape prior to fusion, in order to maximize contact with the opposing cell. Reference Waterman39 Using dynamic imaging of live mouse embryo cultures, Pyrgaki et al. demonstrated that non-neural ectoderm generate filopodia-like extensions that envelop the neural ectoderm and initiate neural tube fusion. Reference Pyrgaki, Trainor, Hadjantonakis and Niswander40 The importance of these filopodia-like extensions are evident in mice genetically deficient in the Ena/VASP family (regulators of filopodia and lamellipodia), as these embryos develop severe NTDs. Reference Menzies, Aszodi and Williams41
The mechanism by which the opposing surfaces adhere and fuse to one another appears to be mediated by several distinct processes. Mice with defects in the ephrin-A5 gene have altered function of the EphA7 tyrosine kinase receptor, which plays a role in balancing cell adhesion and repulsion, show severe NTDs. Reference Holmberg, Clarke and Frisen42,Reference Abdul-Aziz, Turmaine, Greene and Copp43 Altered function of protease-activated receptors PAR1 and PAR2 may also prevent proper fusion of the neural folds, though mice with defective PAR signaling often die in utero due to associated defects. Reference Camerer, Barker and Duong44–Reference Szabo, Hobson, Christoph, Kosa, List and Bugge46 Furthermore, cell adhesion molecules, particularly E-cadherin, serve to hold the neural folds together and facilitate proper neural tube closure. Many of these molecular interactions are regulated by the grainyhead-like (Grhl) transcription factor family. Reference Pyrgaki, Liu and Niswander47,Reference Senga, Mostov, Mitaka, Miyajima and Tanimizu48
Closure of the neural tube is also dependent on a balance between appropriate cell proliferation, differentiation, and programmed cell death. Reference Copp and Greene1,Reference Greene and Copp27,Reference Geelen and Langman49 Anti-mitotic agents are known to increase risk of NTDs, and the association between antifolate medications, or folic acid deficiency and NTDs is thought to be, in part, secondary to the effect on nucleic acid formation and subsequent impaired DNA synthesis. Reference Steegers-Theunissen50–Reference Copp, Brook, Estibeiro, Shum and Cockroft52 Furthermore, studies in mice have revealed that alterations in genes regulating cell cycle progression or cell differentiation can also lead to NTDs, though this occurs more commonly in the cranial region. Reference Harris and Juriloff53 Interestingly, both lack of and excessive apoptosis have been associated with NTDs in animal models. Alterations in apoptotic genes Casp3 and Apaf1 impair closure at the midbrain and hindbrain; Reference Yamaguchi, Shinotsuka and Nonomura54,Reference Juriloff and Harris55 however, excessive cell death has also been associated with failure of neural tube closure, highlighting the delicate balance that must be maintained during neurodevelopment. Reference Greene and Copp27,Reference Fukuda, Tokunaga, Sakamoto and Yoshida56
Closure Sites in Primary Neurulation
Primary neurulation in mammals is discontinuous with the three separate closure locations arising along the developing neural tube, which is in contrast to an older model that describes the neural tube closing in a continuous, zipper-like fashion (Figure 2). Reference Copp, Greene and Murdoch10,Reference Van Allen, Kalousek and Chernoff57 Closure 1 is located at the hindbrain–cervical junction and progresses rostrally and caudally. Failure of this closure leads to craniorachischisis. Reference Copp, Greene and Murdoch10 Closure 2 begins at the boundary of the forebrain and midbrain. It too proceeds both rostrally and caudally. Closure 3 begins at the rostral extent of the forebrain and proceeds caudally. Closures 2 and 3 meet at the anterior neuropore. Closures 1 and 2 join at the hindbrain neuropore. Failure of the neural tube to close anywhere between closures 1 and 3 lead to anencephaly. Both craniorachischisis and anencephaly, which account for about 10% of NTDs, are non-survivable conditions; however, they are quite rare. Reference Greene and Copp27 The rostral extension of Closure 1 terminates at the posterior neuropore. Failure of this process leads to lumbosacral NTDs, the most clinically important entity being MMC. Reference Macdonald, Juriloff and Harris58–Reference Copp and Bernfield60 Although these models are based on studies of mice embryos, there is evidence from clinical cases where patients suffer from multiple simultaneous NTDs that a similar process occurs in humans. Reference Van Allen, Kalousek and Chernoff57,Reference Copp and Bernfield60–Reference Ahmad and Mahapatra62
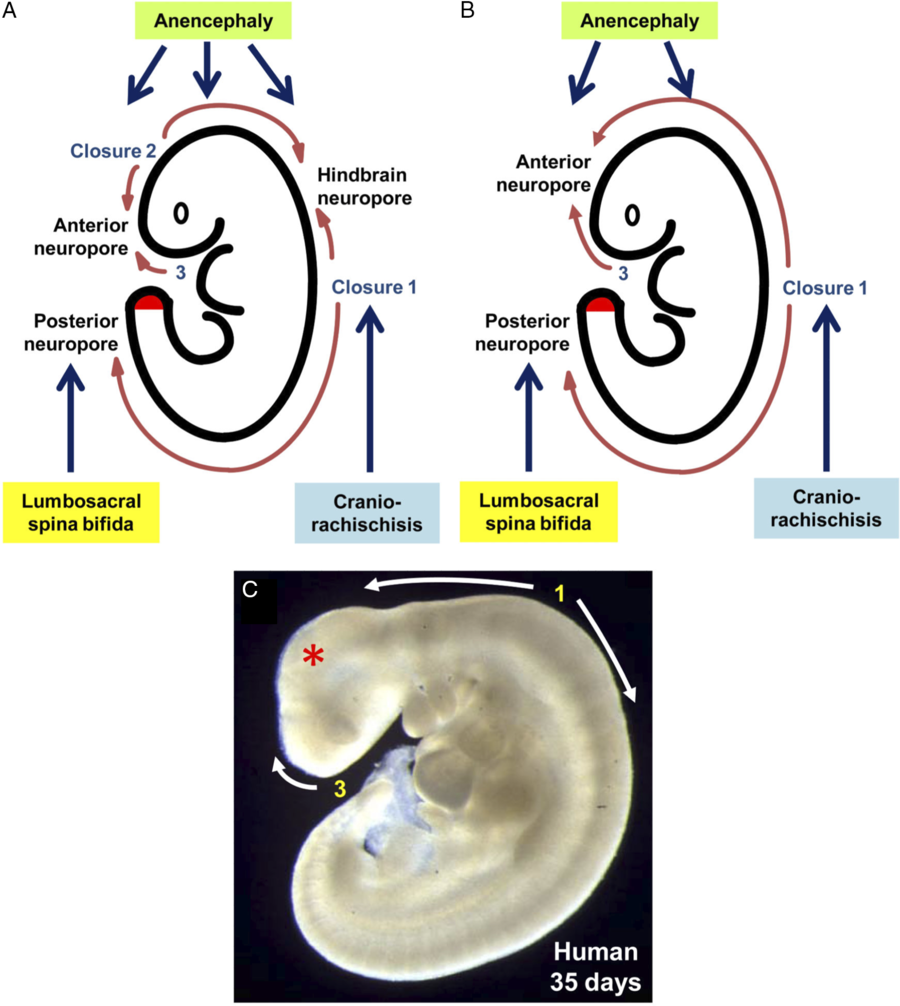
Figure 2: Sites of neural tube closure in mouse and human embryos with the associated NTDs from failure of site closure. Adapted with permission from ref. 1.
Maternal Risk Factors and Folic Acid
As NTDs are severe congenital malformations, there has been a concerted effort to identify modifiable maternal risk factors with the hopes of decreasing the incidence of these birth defects. Of note, maternal obesity and hyperthermia have been linked with elevated rates of NTDs. Reference Waller, Mills and Simpson63–Reference Mitchell67 Nonetheless, the most well-known risk factor for NTDs identified to date is folic acid deficiency. Since large-scale studies demonstrated the association between folic acid supplementation and NTDs, 68,Reference Czeizel and Dudas69 developed countries have seen a dramatic decrease, though not elimination, in NTDs with folic acid fortification of foods. Reference De Wals, Tairou and Van Allen70
The success of folic acid supplementation in reducing the incidence of NTDs has prompted researchers to examine the relationship between genes involved in one-carbon metabolism and neural tube closure. While some studies on mice have identified knockout strains that develop NTDs, Reference Momb, Lewandowski and Bryant71,Reference Narisawa, Komatsuzaki and Kikuchi72 the mechanism behind this relationship remains incompletely understood. Reference Greene and Copp27
Embryological Classification of Different Forms of Spinal Dysraphism
Overview
As described above, normal development consists of a seamless sequence of molecular and cellular events resulting in the formation of the embryo and fetus. For reasons that are not understood, specific steps are vulnerable to failure. There are three key aspects of a dysraphic condition that impact the observed clinical disability: timing during development, the segment involved, and the degree to which the spinal cord is disrupted. Separating different conditions into embryologic categories allows one to anticipate the impact of the condition, and a possible treatment plan.
In general, the earlier the failure occurs in development, the more significant the effect on function. Failures of primary neurulation, which include MMC, occur at very early times in development, probably during the end of the third week after conception, Reference Greene and Copp27,Reference Wood and Smith73 and have profound, deleterious effects on the formation and function of the affected area. Conversely, a dermal sinus tract which occurs after closure of the neural tube is rarely associated with severe neurologic deficits. Reference Ackerman and Menezes74
Like all chordates, mammals have a segmental body plan with a repetitive pattern occurring in the rostral to caudal direction. Specialization of individual segments has occurred with respect to the head and limbs, but the basic segmental plan is clearly seen in the structure of the spine and the innervation of sensory dermatomes. Aside from complete failure of neural tube formation (craniorachischisis), most dysraphic states are restricted to either one or a handful of segments. The number of segments involved and where the affected segments are located along the rostral-to-caudal axis has a direct impact on the patient’s function.
Finally, the degree to which the spinal cord is distorted or affected by the developmental abnormality usually correlates with the severity of the clinical presentation. This is most clearly seen in the diversity of lesions identified as spinal cord lipoma. Understanding the individual contribution of these three factors facilitates both an understanding of the problem and developing a treatment strategy for that patient.
Nomenclature
Dysraphic conditions have been observed for hundreds of years and have acquired various names and terms that are often confusing and do not help in understanding the embryologic origin of these conditions. Reference Rickham75 For example, the term spina bifida aperta is a historical term that referred to “open” NTDs such as MMC but also is considered to include those defects associated with a lack of closure of the epithelial ectoderm. Spina bifida occulta broadly refers to situations where the epithelium was closed over the lesion, but some use the term only to refer to a failure of fusion of the vertebral lamina. These terms are imprecise and should be abandoned.
The only advantage of separating dysraphic conditions into “open” and “closed” forms is that the secondary anomalies that occur in children with MMC (e.g. Chairi II malformation, hydrocephalus, and developmental brain anomalies) rarely occur with lesions that have intact skin. Reference Stoll, Alembik and Dott76,Reference Gilbert, Jones, Rorke, Chernoff and James77
Anomalies of Primary Neurulation
Anencephaly, Craniorachischisis, and MMC
Depending on the specific timing and location, failure of primary neurulation leads to open NTDs such as anencephaly, craniorachischisis, or MMC. Reference Copp and Greene1 In contrast to anencephaly and craniorachischisis, most infants with MMC survive and will progress to a full-term gestation. In mid-gestation fetuses with MMC, the neural tube is clearly visible as a flat structure with a visible neural groove but without recognizable folds present (Figure 3). It is not clear whether the failure of further development of the neural tube is the primary defect leading to lack of normal folding, or whether failure of folding leads to an arrest of subsequent development.

Figure 3: Intrauterine view of MMC demonstrating the flat neural placode with a visible neural groove. Note the absence of a sac that is present when MMCs are repaired postnatally.
MMC is the most common type of open NTD and by the criteria noted above has the most severe (survivable) clinical presentation; the embryologic defect occurs early, it spans multiple segments, and the spinal cord is severely disrupted. The principles of surgical repair of MMC include an anatomic repair of the exposed neural tube (the “placode”) with a multi-layer closure of the dura and skin with the goal of preventing meningitis. There are no techniques available at this time to restore function of the affected nervous system segments. Regardless of whether the placode is closed (“tubularized”) or not, it remains in a low position and usually attaches to the overlying soft tissues. Virtually, all children with MMC will have radiologic evidence of a tethered spinal cord, although generally children do not require surgical release unless they develop new or progressive symptoms. Reference Herman, McLone, Storrs and Dauser78
While the initial repair and subsequent untethering procedures are focused on the site of the open neural tube, the majority of surgical interventions for patients with MMC occur in response to the treatment of the secondary anomalies such hydrocephalus, the Chiari II malformation, urologic dysfunction, and orthopedic deformities.
The relationship between the primary and secondary anomalies observed in MMC is best illustrated by the results of a prospective, randomized clinical trial (the Management of Myelomeningocele Study, or MOMS) which evaluated the efficacy of fetal repair of MMC. Reference Adzick, Thom and Spong79 That study clearly showed that while there was a modest improvement in lower extremity function which is directly related to the spinal cord abnormality, the more significant improvement occurred in the number of children requiring a shunt, and the reduction in the Chiari II malformation. These results support the hypothesis that McLone and Knepper proposed which suggested that an open posterior neuropore allows for persistent cerebrospinal fluid (CSF) leakage into the amniotic fluid, preventing the normal distention of the ventricular system. Reference McLone and Knepper80
Split Cord Malformation
SCMs refer to those anomalies in which an anatomic structure divides two hemi-cords. SCMs are divided into Type 1 and Type 2 based on whether they are housed in separate, or common dural sheaths. In Type 1 SCM, where paired hemi-cords are confined to separate dural sheaths, there is an associated bony or cartilaginous spur separating the two dural sacs. Reference Dias and Pang81 The embryology of SCM is best described by the “unified theory” proposed by Pang et al. According to his hypothesis, SCM arises due to a persistent, accessory neurenteric canal. The theory states that an accessory neurenteric canal leads to a persistent communication between the yolk sac and the amnion, which causes a splitting of the primitive disk and neural plate. Reference Pang, Dias and Ahab-Barmada82 Although SCMs are believed to arise when neurulation is occurring, they differ from MMCs in that the ectoderm is usually closed, and therefore, a skin defect is not present.
The clinical presentation of SCM varies depending upon the age at diagnosis. In newborns and infants, the condition is often discovered because of midline cutaneous abnormalities, such as hypertrichosis, a hemangioma, or a lipoma, or the development of a progressive neurologic deficit. Older children and adults may also be diagnosed while investigating cutaneous stigmata, but oftentimes develop manifestations of tethered cord syndrome leading to the discovery of SCM. Reference Schijman83 Although it has been reported that patients can remain asymptomatic from their SCM, Reference James and Lassman84 it is often suggested that SCMs coming to medical attention should be surgically treated. Reference Schijman83 Repair of Type 1 SCM often involves resecting the bony spur and sectioning of the hypertrophied filum terminale. Repair of Type 2 SCM is directed at releasing the hemi-cords from the fibrous attachments. Reference Pang85
Anomalies of Premature Dysjunction
Spinal Cord Lipomas
Dysjunction is the separation of the neural tube from the overlying neuroectoderm. After the neural tube closes and falls away from the overlying ectoderm, the adjacent mesodermal cells can enter the newly created space and eventually differentiate into dermis, muscle, and bone. Reference Kaufman14 The specific embryologic problem with premature dysjunction is that the neural tube separates from the overlying ectoderm prior to neural tube closure. Reference Morioka, Murakami and Shimogawa86 This is usually segmental, sometimes 1, 2 or 3 segments. The open neural tube allows primitive mesodermal cells to invade the neural tube and these cells ultimately differentiate into fat (Figure 4). Of note, there is a wide range of anomalies that involve lipomas of the spinal cord.
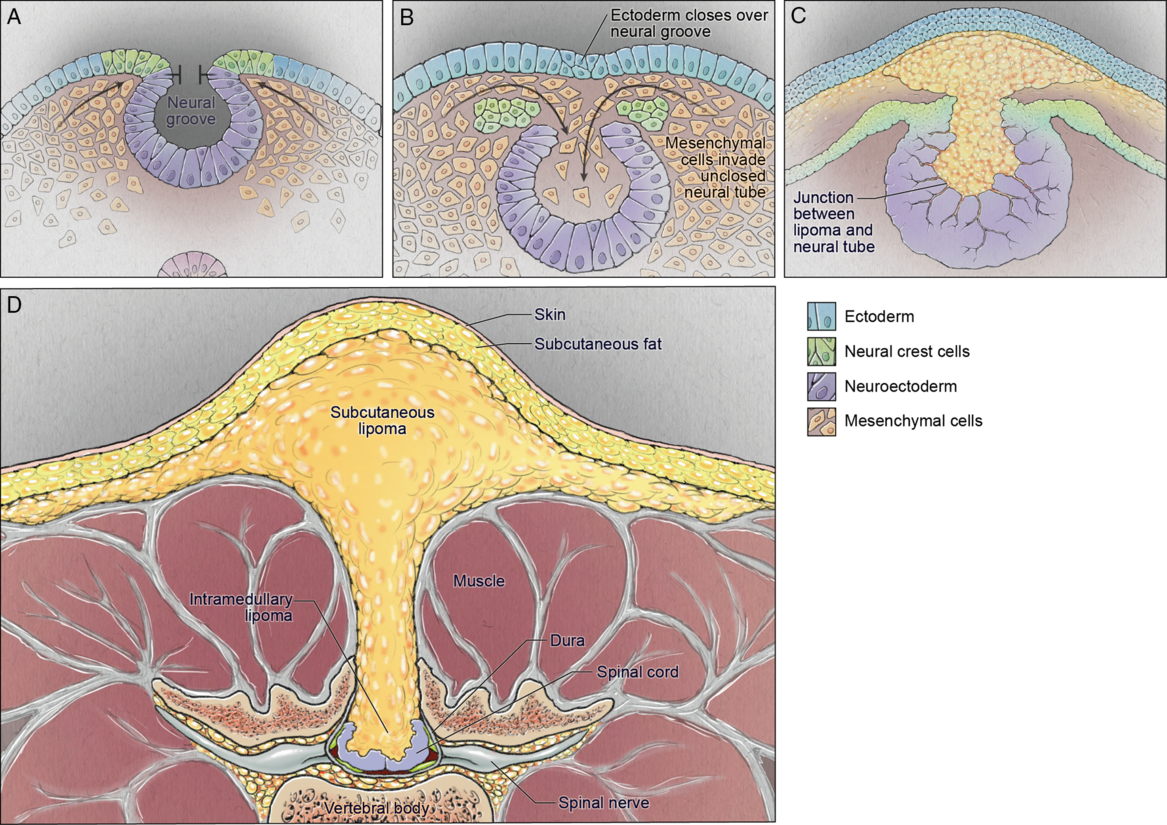
Figure 4: Schematic demonstrating the pathophysiology of spinal cord lipoma/lipomyelomeningocele development: neural folds nearing the point of closure (top left); premature dysjunction of the still open neural tube from the surface ectoderm with mesenchymal cells infiltrating the neural tube (top right); differentiation of mesenchymal cells into fat (bottom left); resultant spinal lipoma (bottom right).
In the simplest form, the neural tube has almost entirely closed prior to dysjunction, and there is small lipoma that extends from the subcutaneous tissues through a fascial and dural defect, and then ascends in the intradural compartment to terminate on the dorsal surface of the spinal cord at the exact neural segment corresponding to the level where the mesodermal defect exists (Figure 5). For example, if the lipoma enters through a fascial defect at L5, it will terminate at the L5 level of the spinal cord. That level of the spinal cord can be in a normal anatomic position, or it can be lower. As the spinal cord is usually normal in structure, most of these patients will have normal or near-normal function. The surgical treatment is generally straightforward with excision of the lipoma.

Figure 5: T1 sagittal magnetic resonance imaging (MRI) demonstrating the segmental nature of a spinal lipoma.
More complicated forms will consist of a lipoma that enters through a multi-segment dorsal defect and then terminate in a diffuse location at the end of the spinal cord. In some cases, the lipoma fills the entire spinal canal and causes a marked distortion of the appearance of the distal spinal cord (Figure 6). For obvious reasons, these lipomas can be difficult to treat surgically since the junction between the cord and the lipoma is often poorly defined and the distal nerve roots can extend through the lipoma. The clinical presentation of patients with these types of lipomas is generally more severe, but inconsistent as some patients with very complex spinal cord lipomas can have minimal symptoms. Asymmetric muscle bulk, weakness, and urologic dysfunction are common. Reference Morioka, Murakami and Shimogawa86

Figure 6: T1 sagittal MRI demonstrating a complex spinal lipoma arising from multi-level mesodermal defect and causing marked distortion of the distal spinal cord with intermixed nerve roots.
Spinal Cord Lipoma and Lipomyelomeningocele
The nomenclature of these entities is applied imprecisely. Strictly speaking, when the lipoma extends into the spinal canal, and the spinal cord is also within the canal, and there is no cystic dilatation of the thecal sac, then these anomalies should be referred to as spinal cord lipomas. If the spinal cord extends beyond the spinal canal into the dorsal soft tissues and there is a cystic component continuous with the subarachnoid space, then these anomalies should be referred to as “lipomyelomeningoceles”. True lipomyelomeningoceles are complex anomalies and probably represent an earlier separation from the ectoderm than spinal cord lipomas. Reference Harley Rhodes87
Surgical Treatment of Spinal Cord Lipomas
An anatomic designation of spinal cord lipomas separates them into dorsal, terminal, or transitional types depending upon the relationship of the lipoma to the spinal cord. Reference Chapman88,Reference Pang, Zovickian and Oviedo89 In general, this mostly reflects the segmental distribution of the original defect. For example, if premature dysjunction occurred at the L5 level, this will lead to a lipoma terminating in the L5 segment of the spinal cord with a “dorsal” configuration. Dysjunction at the lower sacral levels will lead to lipomas with a “terminal” configuration.
Surgery may be indicated for these lesions when they cause tethered cord syndrome. The general surgical principles of partial removal of the lipoma and separation of the spinal cord from the overlying structures remain the same, but with complex forms, it is often impossible to completely separate the spinal cord. There are several features that will predict whether the surgical procedure can be performed with acceptable risk. First is whether there is a discrete, anatomic junction between the lipoma and the spinal cord. If this is present, then complete separation is usually possible (Figure 7). Second is the presence of an identifiable CSF space ventral and preferably lateral to the spinal cord and lipoma (Figure 8). If this is the case, the nerve roots are usually directed in a ventral direction and are not within the lipoma. Third is the presence of a discrete dural entry point of the lipoma (also seen in Figure 8). If present, then the extradural and intradural components are easily separated and disconnected. Conversely, when a lipoma fills the thecal sac without an identifiable ventral CSF space, the lumbosacral nerve roots will often run through the lipoma. This will sometimes prevent adequate untethering of the spinal cord.

Figure 7: A) Intraoperative image demonstrating a discrete tissue plane between spinal lipoma and distal spinal cord; B) complete separation of spinal cord from lipoma along defined anatomic plane.

Figure 8: A and B) T1 and T2 sagittal view MRIs demonstrating a CSF space anterior to the spinal cord along with discrete dural entry point of spinal cord lipoma.
Failure of Primary Dysjunction
The classic anomaly of a failure of primary dysjunction is the dermal sinus tract (Figure 9). It arises from a persisting connection between the neuroectoderm and the ectoderm, typically after the neural tube has closed. Reference Acharya, Pendharkar, Varma, Pruthi and Varadarajan3,Reference Kaufman14,Reference Radmanesh, Nejat and El Khashab90 This abnormality occurs late in neurulation, and it usually involves only one segment and the gross structure of the spinal cord is usually normal. For these reasons, most patients will have either no symptoms or mild symptoms. Although in some cases the tract will contain an actual true lumen lined with epithelium, this is not always the case. When these patients do present to medical attention, it is typically because of tethered cord syndrome or infectious complications, such as meningitis. Reference Radmanesh, Nejat and El Khashab90,Reference Jindal and Mahapatra91 The surgical treatment of dermal sinus tracts is complete excision which can usually be accomplished successfully with low morbidity.

Figure 9: A) T2 sagittal MRI demonstrating persistent dermal sinus tract; B) surgical excision of dermal sinus tract.
Failure of primary dysjunction may also result in limited dorsal myeloschisis (LDM), in which a closed midline skin defect covers, and is connected to, a fibroneural stalk originating from the cord. Reference Pang, Zovickian, Oviedo and Moes92 LDM, which is a rare entity, can be classified as saccular or non-saccular based on its appearance. Reference Pang, Zovickian, Wong, Hou and Moes93 Clinically, LDM presents with tethered cord syndrome detethering is accomplished by sectioning the fibroneural stalk flush with the cord, taking care to protect the functional neural elements. Reference Pang, Zovickian, Oviedo and Moes92,Reference Morioka, Suzuki and Murakami94 Historically, LDM was a common cause of recurrent tethered cord in patients who were diagnosed with meningoceles as the fibroneural stalk was not dealt with. However, with the advent of modern MR imaging and a better understanding of the condition, this is less common. Reference Chatterjee and Rao95
Anomalies of Secondary Neurulation
The CCM is a poorly defined region of tissue that results in the formation of the caudal segments of the spinal cord; typically, the lower sacral segments. These anomalies result in significant problems with urologic function but are otherwise limited in terms of their neurologic impact. Complete failure of formation of the CCM will lead to sacral agenesis and absence of the segments of the conus medullaris below S1. Reference Moore and Lowe96 Associated sacral abnormalities are common. More complex anomalies are those that involve the distal spinal cord and the cloacal organs with lesions such as bladder extrophy, or a combined bladder and vagina.
Anomalies of Post-neurulation Development
Once the neural tube closes, there are abnormalities that can result in significant dysfunction. These include encephaloceles which are a diverse collection of anomalies that occur in different regions of the brain and cranium. Reference Posnick and Costello97 A consistent feature of virtually all encephaloceles is the presence of a normal epithelial layer and a closed neural tube. For this reason, the embryologic origin is felt to be post-neurulation and arising from a failure of mesoderm to form properly over the closed neural tube.
In most situations, the closed neural tube herniates into a skin-covered lesion outside the cranial vault (Figure 10). The ventricle is usually part of the lesion, and the brain within the encephalocele is often dysplastic and nonfunctional. Surgical treatment of an encephalocele requires excision and closure of the sac with truncation of the dysplastic neural tissue if it is felt to be nonfunctional. The degree of brain involvement, and the presence of hydrocephalus, usually determines the patient’s functional outcome.

Figure 10: T1 sagittal MRI demonstrating a posterior fossa encephalocele with ectopic cerebellar tissue herniating into the sac.
Conclusion
Spinal dysraphism is a spectrum of congenital malformations that have a direct relationship to alterations in embryological development. Improving treatment and prevention of these conditions requires clinicians to familiarize themselves with the related neuroembryology. While the past few decades have seen a tremendous advancement in our understanding of these congenital malformations, much remains to be discovered.
Disclosures
The authors have no conflicts of interest to declare.
Statement of Authorship
ME and NG were involved in developing the concept for the paper, compiling the data, and writing the manuscript.










