


Although cerebral vasospasm can rarely complicate traumatic subarachnoid hemorrhage (SAH), ruptured vascular malformations, hemorrhagic brain tumors, and indeed any condition that results in extensive bleeding into the subarachnoid space beneath the brain, in clinical practice vasospasm is by far most commonly associated with rupture of cerebral aneurysms within the basal subarachnoid cisterns. The pathogenesis, diagnosis, and management of vasospasm following cerebral aneurysm rupture are the subjects of this review.
Angiographic vasospasm is arterial narrowing seen on vascular imaging; it begins several days after SAH and peaks in severity about 1 week later. Clinical or symptomatic vasospasm is narrowing causing cerebral ischemia with corresponding symptoms and signs and is sometimes referred to as delayed ischemic neurological deficit. Progression to cerebral ischemia depends largely on the degree and distribution of arterial narrowing. Vasospasm can be focal or diffuse in distribution and mild, moderate, or severe in degree. Vasospasm affects only the intradural arteries and primarily, but not exclusively, arteries and arterioles located on the surface of the brain.
This review will touch upon a more recent suggestion that pathophysiological processes initiated by early brain injury related to aneurysm rupture, and separate from vessel narrowing and cerebral ischemia, may also contribute to delayed neurological deterioration. Determining the exact pathogenesis of this complex condition and developing a corresponding way of preventing it remain ongoing pursuits of neurosurgical research.
Epidemiology of Vasospasm
Angiographic vasospasm is common after rupture of an aneurysm, with an overall incidence of 50-90%.Reference Dorsch and King 1 Rough estimates are as follows: moderate or more severe vasospasm in at least one cerebral artery will develop in two-thirds of patients with ruptured aneurysms, half of these patients will become symptomatic as a result of ischemia, and a cerebral infarct will develop in about half of these symptomatic patients. An analysis of 2741 patients entered into SAH trials in the 1990s found that cerebral infarction had developed in 26% at 6 weeks, which correlated strongly with poorer outcome.Reference Fergusen and MacDonald 2 Cerebral infarction was significantly associated with increasing patient age, worse neurological condition on admission, a history of hypertension or diabetes mellitus, larger aneurysm, induced hypertension as part of management, fever, and a diagnosis of symptomatic vasospasm. With modern SAH management routines, the combined risk for death and permanent disability from vasospasm alone has been reduced to less than 10%, but it still remains one of the leading causes of preventable poor outcome after rupture of an aneurysm.Reference Kassell, Tomer, Haley, Jane, Adams and Kongable 3 - Reference Proust, Hannequin, Langlois, Freger and Crissard 5
Prediction of Vasospasm
A large volume of persistent subarachnoid clot is the most important risk factor predictive of vasospasm after SAH.Reference Friedman, Goerss and Meyer 6 , Reference Reilly, Amidei, Tolentino, Jahromi and MacDonald 7 The original Fisher grading scale in which clot volume and distribution on admission computed tomography (CT) are related to risk for vasospasm has been modified and in one single-center study found to have greater predictive value for delayed ischemia and prognosis.Reference Kramer, Hehir and Nathan 8 This modified Fisher scale (Table 1) scores hemorrhage noted on CT from 0 to 4: 0, no SAH or intraventricular hemorrhage (IVH) (very low risk for vasospasm); 1, focal or diffuse thin layer of SAH, no IVH (low risk for vasospasm); 2, focal or diffuse thin layer of SAH, IVH present (moderate risk for vasospasm); 3, focal or diffuse thick layer of SAH, no IVH (high risk for vasospasm); and 4, focal or diffuse thick layer of SAH, IVH present (highest risk for vasospasm). A slower rate of subarachnoid clot clearance has also been shown to be an independent predictor of vasospasm, although this is not an easy measurement in clinical practice.Reference Reilly, Amidei, Tolentino, Jahromi and MacDonald 7
Table 1 Modified Fisher scale and risk for vasospasm

Other risk factors for the development of vasospasm have been identified, including a loss of consciousness at the time of rupture, poor neurological grade on admission, cigarette smoking, diabetes mellitus or hyperglycemia, and preexisting hypertension (Table 2).Reference Harrod, Bendok and Batjer 9 - Reference de Rooij, Greving, Rinkel and Frijins 11 Premorbid left ventricular hypertrophy in either smokers or hypertensive patients was especially predictive of severe vasospasm in one large single-center series.Reference Inagawa, Yahara and Ohbayashi 12 Factors studied and found to have a possible relationship with risk for vasospasm include female gender, younger patient age, and aneurysm location. A recent single-center review found that distal anterior cerebral artery aneurysms had an unusually high incidence of symptomatic vasospasm.Reference Abla, Wilson and Williamson 13 There has been a suggestion that Japanese people may be more susceptible to vasospasm.Reference Mocco, Ransom and Komotar 14 Cocaine use may also be an independent risk factor for vasospasm.Reference Conway and Tamargo 15
Table 2 Risk factors for vasospasm

Spontaneous perimesencephalic or prepontine SAH (sometimes referred to as “pretruncal” SAH) unassociated with aneurysm rupture is typically a low-volume hemorrhage that clears quickly with a low risk for the development of vasospasm.Reference Schwartz and Solomon 16 However, thick and persistent nonaneurysmal SAH of this type is associated with a risk for vasospasm.
There is some evidence that endovascular coiling, as opposed to microsurgical repair of ruptured aneurysms, is associated with a lower risk for the subsequent development of vasospasm,Reference Rabinstein, Pichelmann and Friedman 17 - Reference Ibrahim, Morgan and Macdonald 19 although a rigorous comparison has not yet been made. Intraoperative aneurysm rupture during surgical clipping was not found to correlate with increased vasospasm in a large single-institution review.Reference Sheth, Hausrath, Numis, Lawton and Jospehoson 20 Impaired cerebral autoregulation assessed by transcranial Doppler ultrasound techniques was found to be predictive of vasospasm when detected within the first few days following SAH in a recent single-center study.Reference Orite, Mink and Tan 21
Pathogenesis
Smooth Muscle Contraction
Vasospasm is prolonged cerebral arterial constriction caused by vascular smooth muscle contraction. The hemoglobin released from subarachnoid blood clots triggers the entry and release of calcium and subsequent activation of calcium/calmodulin-dependent myosin light-chain kinase, which in turn leads to phosphorylation of the myosin light chain and induces actin and myosin cross-linkage and mechanical shortening (smooth muscle contraction). Such contraction requires adenosine triphosphate and calcium, and vascular smooth muscle relies more on extracellular than intracellular calcium stores, which enter through voltage-gated and receptor-operated calcium channels. Although myofilament activation depends on calcium and high-energy phosphates, chronic vasospasm, which ensues days later and lasts up to several weeks, does not. The contractile proteins protein kinase C, Rho kinase, and protein tyrosine kinase and their corresponding signal transduction pathways have been implicated in vasospasm models when their activation shifts the contractile mechanism toward increased shortening in the absence of high intracellular calcium levels.Reference Koide, Nishizawa, Ohta, Yokoyama and Namba 22 , Reference Hansen-Schwartz, Vajkoczy, Macdonald, Pluta and Zhang 23 This contiguous and second-phase “chronic” vasospasm is less reversible with pharmacologic vasodilators both in animal modelsReference Megyesi, Vollrath, Cook, Chen and Findlay 24 and in humans undergoing intra-arterial, pharmacologic vasodilation treatment.
Sustained vasoconstriction is associated not only with functional impairment of the vessel, but also with ultrastructural damage to the vascular wall layers, including vacuolization of endothelial cells and loss of tight junctions, breakage of the internal elastic lamina, and patchy myonecrosis in the tunica media.Reference Findlay, Weir, Kanamaru and Espinosa 25
Endothelial Injury, Nitric Oxide, and Endothelin-1
Autooxidation of the oxyhemoglobin contained in blood clots encasing cerebral arteries produces methemoglobin and superoxide anion radical, which in turn lead to lipid peroxidation.Reference Sasaki, Wakai, Asano, Watanabe, Kirino and Sano 26 , Reference Kamezaki, Yanaka, Nagase, Fujita, Kato and Nose 27 Harmful hydroxyl radicals and lipid peroxides permeate the vessel wall and injure endothelial and smooth muscle cells.Reference Ohta, Satoh and Kuroiwa 28 , Reference Foley, Takenaka, Kassell and Lee 29 Damage to the endothelium in particular is thought to play a key role in the establishment of vasospasm, either through the loss of endothelial nitric oxide (NO) synthesis, an important vasodilator and regulator of vascular tone, or through the overproduction of endothelin, a powerful vasoconstrictor peptide.Reference Iuliano, Pluta, Jung and Oldfield 30 These two endothelial-derived substances and the possible imbalance in their production after SAH are at the center of experimental vasospasm research at the present time.
Decreased availability of the simple molecule NO may contribute to the development of vasospasm; supporting this hypothesis are the following observations following experimental SAH in animals: (1) endothelial NO synthase (eNOS) dysfunction in vasospastic vessels, (2) NO scavenging by oxyhemoglobin, (3) reversal of vasospasm by NO donors, (4) disappearance of neuronal NO synthase activity from the adventitia of vasospastic vessels, and (5) decreased cerebrospinal fluid nitrite levels along with increased levels of asymmetric dimethyl-l-arginine, the endogenous inhibitor of NO synthase.Reference Moon, Gajdusek, London and Mayberg 31 - Reference Jung, Oldfield and Harvey-White 39
Endothelin-1 (ET-1) is the predominant isoform of endothelin and has the greatest role in vasoconstriction. ET-1 is a 21-amino acid cleavage product of a 212-amino acid peptide precursor, the final step mediated by endothelin-converting enzyme. It is released on the abluminal side of the tunica media, acts on neighboring vascular smooth muscle endothelial receptor A (ETA) receptors, and causes profound and sustained vasoconstriction.Reference Chow, Dumont and Kassell 40 ET-1 levels have been found to be elevated in the cerebrospinal fluid of patients in whom vasospasm and brain ischemia develop,Reference Seifert, Löffler, Zimmermann and Stolke 41 - Reference Mascia, Fedorko and Stewart 43 and either inhibition of ET-1 production or antagonism of its effect has prevented vasospasm in animal models.Reference Chow, Dumont and Kassell 40
It seems possible that derangements in either or both NO or ET-1 metabolism by cerebral vascular endothelium may play a critical role in the pathogenesis of vasospasm.
Inflammation, Vessel Remodeling, and Vasospasm
Although it has become clear that vasospasm is not a type of vasculitis, there is evidence that inflammatory mechanisms are activated after SAH and may therefore be involved in the development of vasospasm, possibly by contributing to vasocontraction or modifying the vessel wall extracellular matrix and smooth muscle cell phenotype—a process known as vascular “remodeling.”Reference Dumont, Dumont and Chow 44 Inflammatory cytokines,Reference Sasaki, Kasuya and Onda 45 , Reference Bowman, Dixit, Bonneau, Chinchilli and Cockruft 46 intercellular adhesion molecules,Reference Clatterbuck, Gailloud and Ogata 47 , Reference Pradilla, Wang, Legnani, Frazier and Tamargo 48 and genetic upregulation of inflammatory, proliferative, and extracellular matrix–regulating genesReference Macdonald, Zhang, Ono and Komuro 49 - Reference Vikman, Ansar and Edvinsson 51 have been examined. In humans, elevations of plasma complement C3a and soluble intercellular adhesion molecule-1 have been associated with poor outcome and the development of vasospasm, respectivelyReference Mack, Ducruet and Hickman 52 , Reference Mocco, Mack and Kim 53 ; increased intrathecal levels of the cytokine interleukin-6 predicted vasospasm in another small study.Reference Schoch, Regel, Wichert, Graser, Vollbracht and Stolke 54
Although the precise pathogenesis of vasospasm is still the subject of investigation, a prolonged, biphasic vasoconstrictive process in which the second, chronic phase is mechanistically distinct from the first seems most consistent with what has been seen experimentally and in humans. It remains possible that additional processes, including inflammation, may contribute to the pathogenesis of this condition (Table 3).
Table 3 Theories on the pathogenesis of vasospasm

Early Brain Injury
Vessel narrowing has traditionally been considered the major cause of delayed-onset neurological deterioration following SAH, although it is clear that brain condition, the status of anatomical collateral blood supply, and systemic factors including circulating blood volume, blood pressure, and hematocrit importantly influence the impact of vasospasm on patients as it develops. The sometimes seen dissociation between location and severity of arterial narrowing and territories of cerebral ischemiaReference Brown, Kumar, Dhar, Samson and Diringer 55 and the failure of drugs targeting vasoconstriction to improve outcomeReference Etminan, Vergouwen, Ilodigwe and MacDonald 56 has shifted attention to other possible but not yet recognized mechanisms behind delayed neurological deterioration after SAH. Early brain injury resulting from a spike in intracranial pressure and global cerebral ischemia at the time of aneurysm rupture may trigger a cascade of pathological processes, including inflammation, cortical spreading depression, capillary thrombosis, blood–brain barrier dysfunction, cerebral edema, and neuronal apoptosis.Reference Rowland, Hadjipavlou, Kelly, Westbrook and Pattinson 57 - Reference Sanchez-Porras, Zheng, Santos, Scholl, Unterberg and Sakowitz 60 In combination with vasospasm, one or more of these processes may also underlie delayed neurological worsening after aneurysmal SAH.
Clinical Features and Investigation
Symptoms, Signs, and Differential Diagnosis
Coincident with its delayed onset, symptoms of ischemia resulting from cerebral vasospasm most commonly appear 1 week after aneurysm rupture but often occur later; accordingly, it is important to remain vigilant for this complication for at least 2 weeks after SAH. Regular and careful bedside examination remains the simplest and most effective means of detecting early ischemia in awake, examinable patients; one should concentrate on subtle findings such as diminished attention, changes in verbal output, or a new pronator drift of the upper extremity. Symptomatic vasospasm usually has a gradual onset, sometimes heralded by increased headache and either agitation or somnolence that is a change in patient behavior. It then follows a progressive course if untreated. A smaller group of patients will experience precipitous deterioration.Reference Fisher, Roberson and Ojemann 61 Signs of symptomatic vasospasm are referable to the territory that has become ischemic and are most easily distinguished when they lateralize to a middle cerebral artery (MCA) territory with monoparesis or hemiparesis and, when the dominant hemisphere is affected, aphasia. Anterior cerebral artery vasospasm can be marked by leg weakness, sometimes bilateral in distribution, as well as confusion, drowsiness, poverty of speech, and eventually abulia. Vertebrobasilar vasospasm can cause more generalized neurological deterioration, with a reduced level of consciousness an early sign followed by any pattern of body weakness.
Vasospastic ischemia is difficult to detect in patients who remain in poor neurological condition following SAH, so monitoring of comatose patients with ancillary techniques (discussed later) is particularly important.
Delayed neurological deterioration after aneurysmal SAH has several causes, including increased edema (surrounding intracerebral hematomas, surgical contusions, or infarcts), rebleeding of the aneurysm or an aneurysmal remnant, hydrocephalus, sepsis (including meningitis and ventriculitis), hyponatremia, hypoxia, and hypotension (Table 4). Any one (or a combination) of these conditions can magnify a preexisting neurological deficit and therefore easily be mistaken for vasospasm. Care has to be taken not to automatically assume vasospasm and cerebral ischemia are the causes of delayed neurological deterioration in patients recovering from aneurysmal SAH; a diligent search for other causes must be carried out.
Table 4 Causes of delayed deterioration after subarachnoid hemorrhage

On the other hand, previously mentioned in this review is a small subgroup of patients who suffer a delayed and often global decline in neurological status for which no underlying cause can be identified, including cerebral vasospasm. Early brain injury caused by an acute elevation in intracranial pressure and global cerebral ischemia at the time of aneurysm rupture may trigger a cascade of pathological processes, including cerebral inflammation, cortical spreading depolarization, microthrombosis, blood–brain barrier breakdown, cerebral edema, and apoptosis.Reference Hansen-Schwartz, Vajkoczy, Macdonald, Pluta and Zhang 23 , Reference Rowland, Hadjipavlou, Kelly, Westbrook and Pattinson 57 - Reference Sanchez-Porras, Zheng, Santos, Scholl, Unterberg and Sakowitz 60 , Reference Simard, Aldrich, Schreibman, James, Polifka and Beaty 62
Diagnosis
Diagnosis of symptomatic vasospasm requires that the other causes of delayed worsening listed earlier be ruled out with CT and appropriate laboratory investigations. If vasospasm remains the most likely cause of deterioration and treatment by induced hypertension reverses the deficit, the diagnosis can usually be assumed and additional testing is at the discretion of the treatment team. Failure to respond neurologically in this scenario, as well as evaluation of comatose patients, requires additional testing (Table 5). The following are most commonly used monitoring methods after SAH.
Table 5 Diagnosis of vasospasm
Transcranial Doppler
Transcranial Doppler (TCD) works on the principle that, as an artery narrows, blood flow velocity within it increases, causing a Doppler shift frequency between emitted and reflected waves.Reference Purkayastha and Sorond 63 TCD is frequently used to monitor patients with SAH for increases in intracranial blood flow velocity suggestive of incipient vasospasm. TCD examinations are noninvasive, are carried out at the bedside, and can easily be performed on a daily basis providing patients have an adequate acoustic window in their temporal region through which to insonate. TCD is best used to detect proximal vasospasm because of segmental and diffuse increases in blood flow velocity in these vessels compared with more distal vessels.Reference Purkayastha and Sorond 63 There is good general correlation between TCD velocities and vasospasm, with velocities in the MCA greater than 120 cm/second being indicative of some degree of vasospasm and those greater than 200 cm/second consistent with severe vasospasm. Because other factors such as blood pressure and overall cerebral blood flow (CBF) can influence velocity, distinguishing vasospastic from hyperemic increases in blood velocity has been reported to be facilitated by measuring cervical internal carotid artery velocity in addition to intracranial blood velocity.Reference Lindegaard, Bakke, Sorteberg, Nakstad and Nornes 64 A “Lindegaard ratio” of VMCA/VICA greater than 3 is consistent with vasospasm (hyperemia is associated with increased velocity in both the MCA and internal carotid artery, so the ratio is the same).Reference Purkayastha and Sorond 63 , Reference Lindegaard, Bakke, Sorteberg, Nakstad and Nornes 64 A similar velocity ratio between the basilar artery and the extracranial vertebral artery has been proposed to improve the sensitivity and specificity of detecting basilar artery vasospasm.Reference Sviri, Ghodke and Britz 65
The clinical utility of TCD is best when MCA values are clearly low (<120 cm/second) or very high (>200 cm/second), and the respective negative and positive predictive values for significant angiographic vasospasm in the MCA trunk are close to 90%.Reference Vora, Suarez-Almazor, Steinke, Martin and Findlay 66 - Reference Mariak, Krejza, Swiercz, Kordecki and Lewko 68 For values in the intermediate range, additional maneuvers may improve its accuracy in detecting vasospasm, such as testing for hyperemic autoregulatory responses to transient, manual carotid compression,Reference Lam, Smielweski, Czosnyka, Pickard and Kirkpatrick 69 , Reference Ratsep and Asser 70 or combining the TCD information with CBF measurements.Reference Gonzalez, Boscardin, Glenn, Vinuela and Martin 71 TCD does not reliably detect vasospasm in more peripheral branches,Reference Okada, Shima and Nishida 72 which may account for its failure to always correlate with perfusion deficits detected on CBF studies.Reference Minhas, Menon and Smielewski 73 Regular surveillance of SAH patients with TCD ultrasonography by skilled technicians is considered helpful in many neurosurgical units, but the results must be considered in the context of the individual patient along with all other information available.
CBF and Perfusion
Single-photon emission CT,Reference Egge, Sjoholm, Waterloo, Solberg, Ingebrigsten and Romner 74 quantitative stable xenon-enhanced CT,Reference Gonzalez, Boscardin, Glenn, Vinuela and Martin 71 and positron emission tomographyReference Minhas, Menon and Smielewski 73 can be used to detect cerebral ischemia after SAH, but none is a practical investigation that can be easily performed or repeated in critically ill patients. Magnetic resonance imaging to look for perfusion deficitsReference Hertel, Walter, Bettag and Mosdorf 75 or ischemia on diffusion-weighted imagesReference Condette-Auliac, Bracard and Anxionnat 76 , Reference Weidauer, Lanfermann, Raabe, Zanella, Seifert and Beck 77 has the same limitation.
Computed tomographic perfusion (CTP) or “perfusion CT scanning” has been used in patients with SAHReference van der Schaaf, Wermer, van der Graaf, Velthuis, van der Kraats and Rinkel 78 , Reference Sviri, Britz, Lewis, Newell, Zaaroon and Cohen 79 and has rapidly gained popularity because of its ease of use, speed of image acquisition, and good correlation with digital subtraction angiography for angiographic vasospasm.Reference Washington and Zipfel 80 It requires a contrast bolus to calculate regional CBF. Cerebral ischemia can be detected on the basis of side-to-side differences in perfusion or absolute thresholds values for CBF and mean transit time. CTP can provide early detection of reduced CBF, prolongation of mean transit time (which is also indicative of ischemia), and the status of cerebral blood volume, which is normally maintained in penumbral brain, but markedly reduced in established infarcts.Reference Dankbaar, de Rooij and Rijsdijk 81 In a recent meta-analysis, the sensitivity of CTP in identifying patients with angiographic vasospasm was 74% and specificity 93%.Reference Greenberg, Gold and Reichman 82
Thermal diffusion flowmetry has been used as continuous bedside monitoring in SAH patients to detect vasospasm causing significant reductions in CBF.Reference Vajkoczy, Horn, Thome, Muc and Schmiedek 83 Provided that the white matter location into which the thermal diffusion microprobe is inserted (usually the frontal lobe) is representative of the territory at risk, continuous measurement of CBF values could be useful in patients with high-grade SAH who cannot be assessed neurologically. It is an invasive monitor and because only a very small area of brain is being examined, sampling errors and false-negative results are a concern.
Near-Infrared Spectroscopy
Near-infrared spectroscopy (NIRS) may prove to be a useful technology for simple, noninvasive bedside monitoring of cerebral ischemia following SAH.Reference Mutoh, Ishikawa, Suzuki and Yasui 84 - Reference Brawanski, Faltermeier, Rothoerl and Woertgen 86 Probes are secured to the scalp with adhesive pads, allowing for prolonged and continuous monitoring. NIR light (700-1000 m) penetrates superficial layers (e.g. skin, subcutaneous fat, skull) and is either absorbed by chromophores (oxy- and deoxyhemoglobin) or scattered within the tissue. Measurement is based on the ability of light to penetrate the skull and to determine hemoglobin oxygenation according to the amount of light absorbed by hemoglobin. Unlike pulse oximetry (which uses a single sensor), cerebral oximetry with NIRS uses two photodetectors with each light source. The technology allows selective sampling of tissue beyond a specified depth beneath the skin. Near-field photodetection then can be subtracted from far-field detection to provide selective measurements of tissue oxygenation.
NIRS is a noninvasive and relatively low-cost optical technique for measuring tissue oxygen saturation, changes in hemoglobin volume, and, indirectly, CBF. NIRS detects ischemia through a decrease in cortical oxygen saturation and total hemoglobin. Time-resolved NIRS permits quantitative measurements of hemoglobin concentration, and in one pilot study was found more sensitive than TCD in the diagnosis of vasospasm.Reference Yokose, Sakatani and Murata 87 Verification from studies with larger sample sizes is required.
The limitation of NIRS is that the measurements of hemoglobin concentrations are averages, and sample only a small area of cerebral tissue. NIRS measures blood oxygenation predominantly in the venous compartment of a certain cortical region and the cerebrospinal fluid, the skull, and the other layers of the human head can interfere with the photon signals.Reference Yoshitani, Kawaguchi and Miura 88 NIRS probes can only be used on shaven or hairless parts of the head, most commonly the frontal regions. The positive feature of NIRS is its utility as a continuous bedside monitor for cortical blood oxygen saturation, total brain hemoglobin, oxyhemoglobin, and deoxyhemoglobin, which can be helpful in a multimodal approach to management.
Microdialysis Monitoring
Cerebral microdialysis catheters allow continuous bedside measurement of extracellular concentrations of glutamate, lactate, pyruvate, glucose, and glycerol in brain tissue, thereby screening for excitotoxic cell injury characterized by elevations in lactate with respect to glucose and pyruvate levels and an increase in the glycerol concentration. This somewhat demanding monitoring technique provides chemical information about a very small region of the brain, but it has been used, along with cerebral oximetry, to detect ischemia in patients with SAH.Reference Unterberg, Sakowitz, Sarrafzadeh, Bennedorf and Lanksch 89 - Reference Skjoth-Rasmussen, Schulz, Kristensen and Bjerre 91 The markers with the most reliable prediction of ischemia are glutamate and lactate because these peak 24 hours before clinical ischemia followed by glycerol concentrations that peak 12 hours before clinical ischemia. Furthermore, raised lactate/glutamate ratios and lactate/pyruvate ratios are good prognostic indicators in SAH.Reference Spiotta, Provencio, Rasmussen and Manno 92 It should be noted that high lactate by itself does not indicate ischemia, but rather a hypermetabolic state that may show improved outcome.
Brain Tissue Oxygen
Direct cerebral oximetry can be carried out with a commercially available intraparenchymal brain probe (Licox Brain Tissue Oxygen Probe, Integra Neurosciences), which provides the partial pressure of oxygen in a continuous fashion.Reference Spiotta, Provencio, Rasmussen and Manno 92 A level below 10-15 mmHg is considered to represent hypoxia, but as with the microdialysis probe, direct cerebral oximetry is an invasive technique and samples only a minute territory of brain, as is the case with all parenchymal tissue probes.
Vascular Imaging
The most practical method of imaging large- and medium-sized cerebral arteries is digital subtraction angiography or high-definition computed tomographic angiography.Reference Otawara, Ogasawara, Ogawa, Sasaki and Takahashi 93 Magnetic resonance imaging is appealing in terms of the amount of information it can provide, but is difficult to carry out in critically ill patients.Reference Hattingen, Blasel, DuMesnil, Vatter, Zanella and Weidauer 94
Computed tomographic angiography had a collective sensitivity and specificity of 80% and 93%, respectively, in ten recent studies but failed to be as accurate as digital subtraction angiography because of artifacts from clips and coils. It has been recommended as an initial screening test.Reference Washington and Zipfel 80 Catheter-based angiography is best in patients in whom balloon angioplasty is being considered.
Angiographic vasospasm is a concentric narrowing that can be focal, segmental, or diffuse. It is commonly graded as mild (<25%), moderate (25% to 50%), or severe (>50%) in comparison to baseline, prevasospasm imaging. “Early” vasospasm on admission angiography and within 48 hours of aneurysm rupture has been described in a small percentage of SAH patients, although without baseline angiography it is not clear how this diagnosis can be made accurately in all cases.Reference Baldwin, Macdonald and Huo 95 Its detection has been associated with the development of cerebral infarction and poor outcome.
Along with TCD and CBF measurements, properly timed and sometimes-repeated cerebral angiography is important in patients who are in poor neurological condition and in whom physical examination is difficult or inaccurate. If discovered, significant large-vessel vasospasm can then be treated.
Prevention of Vasospasm and Cerebral Protection
General Measures: Fluid Management and Medical Treatment
Patients have a tendency toward volume contraction in the acute stage of SAHReference Nakagawa, Su, Sato and Shirane 96 and hypovolemia should be carefully avoided. It is not clear that a deliberate attempt to induce hypervolemia with volume expansion therapy is beneficial in terms of prevention of vasospasm or ischemia or even possible in patients with normal renal function.Reference Lennihan, Mayer and Fink 97 - Reference Rinkel, Feigin, Algra and van Gijn 100 Patients should be hydrated with at least 3 L of isotonic fluids daily, and there is some evidence that additional colloid infusions (such as albumin) are beneficial.Reference Suarez, Shannon and Zaidat 101 Some SAH patients experience excessive natriuresis known as “cerebral salt wasting” that is usually related to elevations in brain natriuretic peptide, and they are susceptible to the development of hyponatremia during this time.Reference Rahman and Friedman 102 Hyponatremia may increase the risk for vasospasmReference McGirt, Blessing and Nimjee 103 and is associated with cerebral infarction in poor-grade patients.Reference Zheng, Qiui and Jin 104 Importantly, hyponatremia in the setting of SAH should not be treated with fluid restriction but rather with salt replacement in the form of normal or 3% hypertonic saline, combined with fludrocortisone (Florinef) administration (0.3 mg/day) if the patient is experiencing vasospasm.Reference Rahman and Friedman 102 , Reference Mori, Katayama, Kawamata and Hirayama 105
Blood transfusion has been associated with a higher likelihood of both vasospasm and poor outcome in SAH patients,Reference Kramer, Hehir and Nathan 8 , Reference Smith, Le Roux, Elliott and Winn 106 suggesting that anemia is a marker for other factors that contribute to SAH-associated morbidity. The optimal hemoglobin concentration in patients with SAH in terms of hematocrit, blood viscosity, and oxygen delivery to the brain is not known with certainty, but is generally believed to be higher than 9 g/dl.
Systemic blood pressure should be maintained in the normotensive to slightly hypertensive range, provided that the aneurysm has been repaired. In patients with external ventricular drains, cerebral perfusion pressure (CPP) is a more important parameter to monitor and maintain at 70 mmHg or greater in poorer-grade patients.Reference Bailes, Spetzler, Hadley and Baldwin 107
Optimal ventilation and oxygenation, prevention of fever, maintaining euglycemia, good nutrition, and attention to the concentration of all electrolytes in addition to sodium are important in reducing the impact of vasospasm and delayed ischemia.
Nimodipine, administered orally or via a nasogastric tube, 60 mg every 4 hours and continued for 3 weeks (for patients requiring that length of hospitalization), is standard treatment of patients with aneurysmal SAH because it was shown to have a statistically significant beneficial impact on clinical outcome, however modest.Reference Dorhout Mees, Rinkel and Feigin 108 Nimodipine prevents increases in intracellular calcium by blocking dihydropyridine-sensitive (L-type) calcium channels, but its mechanism of effect in patients with SAH is unknown. It does not appear to reduce angiographically detectable vasospasm. It may work as a neuroprotective agent. Nimodipine can cause temporary depression of blood pressure, in which case the dose can be reduced and, if possible, given more frequently (i.e. 30 mg every 2 hours) (Table 6).
Table 6 Prevention of vasospasm and cerebral protection

ICP, intracranial pressure.
Investigational Preventive Treatments
It is beyond the scope of this review to review the countless number of pharmacologic agents that have been tested in experimental models of SAH and vasospasm, many of which, by targeting a specific pathologic process, were intended to explore a particular pathogenetic theory for vasospasm. The following are preventive strategies that have been tested clinically.
Prophylactic Balloon Angioplasty
Based on the observation that angioplasty of canine carotid arteries prevented the development of vasospasm when the arteries were subsequently encased in blood clots,Reference Megyesi, Findlay, Vollrath, Cook and Chen 109 prophylactic transluminal balloon angioplasty was tested in a phase II multicenter, randomized trial.Reference Zwienenberg-Lee, Hartman and Rudisill 110 A total of 170 patients with thick subarachnoid clots were enrolled and 85 were randomized to undergo percutaneous transluminal balloon angioplasty within 96 hours of SAH; the major cerebral arteries targeted included the supraclinoid internal carotid artery, MCA M1 segments, and the basilar artery. Although there was a trend toward reduced vasospasm in the patients treated prophylactically by percutaneous transluminal balloon angioplasty, outcome was no different from that in the control group, and the safety of this treatment was questionable; four patients had a procedure-related vessel perforation, three of whom died.
Clot Clearance
Intracisternal thrombolysis with either recombinant tissue plasminogen activator or urokinase speeds clearance of subarachnoid clots, and there is evidence that this is associated with prevention of vasospasm and less cerebral ischemia (Figures 1 and 2).Reference Amin-Hanjani, Ogilvy and Barker 111 However compelling the rationale of intrathecal thrombolysis, safety concerns about introducing a thrombolytic agent into the subarachnoid space in a recently injured brain has precluded wide acceptance of this approach. Kinetic therapy in the form of “head shaking” with cisternal thrombolysis,Reference Kawamoto, Tsutsumi and Yoshikawa 112 or intraventricular fibrinolysis,Reference Etminan, Beseoglu, Eicker, Turowski, Steiger and Hanggi 113 or with lumboventricular lavageReference Hanggi, Liersch, Turowski, Yong and Stieger 114 has been reported to be beneficial in single-center studies, as has simple lumbar drainage.Reference Klimo, Kestle, MacDonald and Schmidt 115 , Reference Al-Tamimi, Bhargava and Feltbower 116 Finally, one report has suggested that fenestration of the lamina terminalis at the time of aneurysm clipping reduces the incidence of not just subsequent hydrocephalus, but also vasospasm from 55% to 30% in patients with thick subarachnoid clots, the proposed mechanism being increased cerebrospinal fluid flow and clot clearance in the basal subarachnoid cisterns.Reference Andaluz and Zuccarello 117

Figure 1 This 55-year-old man suffered a sudden headache with vomiting during coitus and on arrival to hospital several hours later was responsive with a normal neurological examination. CT scanning showed diffuse, symmetric, and thick SAH (A, Fisher grade 4, very high risk of vasospasm) as well as hydrocephalus. Immediately following clip repair of his anterior communicating artery aneurysm, 10 mg of recombinant tissue plasminogen activator (rt-PA) was administered into the basal cisterns. The following morning, CT scanning showed good clearance of the clot from the central basal cisterns, with some residual hematoma in the left Sylvian fissure (B, arrow; also see Figure 2).
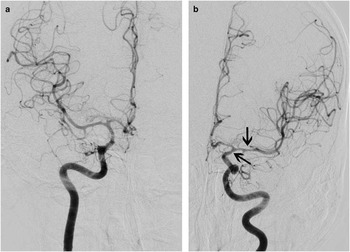
Figure 2 The patient did not experience delayed ischemia, and cerebral angiography 8 days later showed only mild vasospasm of the left supraclinoid internal carotid and proximal middle cerebral arteries (A, anteroposterior projection of the right internal carotid artery injection; B, anteroposterior projection of the left internal carotid artery injection, with arrows).
Intrathecal Vasodilators
Prolonged-release nicardipine implants placed in the subarachnoid space of patients with thick clots and undergoing surgical clipping appeared to significantly prevent vasospasm and ischemia in a single-center, nonrandomized cohort studyReference Kasuya, Onda, Sasahara, Takashita and Hori 118 ; these results are similar to those in a previous study in which papaverine pellets were used.Reference Dalbasti, Karabiyikoglu, Ozdamar, Oktar and Cagli 119 A randomized, placebo-controlled phase IIa trial of prolonged-release nicardipine surgical implants found that nicardipine reduced the incidence of vasospasm and delayed ischemic deficits as well as improving clinical outcome after severe SAH and aneurysm clipping.Reference Barth, Capelle and Weidauer 120 However promising, it is unclear how this form of nicardipine treatment could be applied to patients undergoing endovascular coiling of ruptured aneurysms.
There is interest in administering nimodipine in a slow-release gel to patients via an external ventricular drain to act as a direct vasodilator and possible neuroprotectant, and a pilot study including patients with aneurysms either clipped or coiled is under way (Edge Therapeutics EG-01-1962-02 Nimodipine Microparticles to Enhance Recovery While reducing Toxicity after Subarachnoid Hemorrhage study).
Magnesium
Magnesium sulfate (MgSO4) has neuroprotective and vasodilatory properties and has therefore been tested for the prevention of vasospasm and ischemia in patients with SAH. One early study found no benefit from intravenous MgSO4 infusions,Reference Veyna, Seyfried and Burke 121 another showed various trends toward benefit,Reference Stippler, Crago and Levy 122 and a third suggested efficacy equivalent to nimodipine in preventing ischemic damage.Reference Schmid-Elsaesser, Kunz, Zausinger, Prueckner, Briegel and Stieger 123 Several additional trials have since been completed, including several large, international, and multicenter phase 3 randomized controlled studies, Intravenous Magnesium Sulfate for Aneurysmal Subarachnoid Hemorrhage (IMASH; 327 patents enrolled) and IMASH-2 (1204 patents enrolled).Reference Wong, Chan, Boet, Poon and Gin 124 , Reference Mees, Algra and Vendertop 125 Those large trials, as well as a recent meta-analysis, found that intravenous magnesium (20 mmol MgSO4 administered over 30 minutes followed by a continuous infusion of 80 mmol MgSO4 per day for 14 days) did not increase the probability of good neurological outcome or decrease the risks of cerebral infarction, radiographic vasospasm, or mortality.Reference Golan, Vasquez, Ferguson, Adhikari and Scales 126 There is no evidence at this time to support the use of intravenous magnesium sulfate following aneurysmal SAH.
Endothelin Receptor Antagonists
As discussed earlier, ET-1 overproduction from cerebral endothelial cells damaged by SAH is one of the leading theories behind the pathogenesis of cerebral vasospasm. The ETA/B receptor antagonist TAK-044 was found to reduce delayed ischemia with an acceptable safety profile in a phase II randomized controlled trial, but with no apparent effect on outcome.Reference Shaw, Vermeulen, Murray, Pickard, Bell and Teasdale 127 The ETA receptor antagonist clazosentan, which showed promising results in a multicenter phase IIa study,Reference Vajkoczy, Meyer and Weidauer 128 has now been studied in a randomized, double-blind, placebo-controlled, phase II dose-finding trial (Clazosenatan to Overcome Neurological iSChemia and Infarct OccUrring after Subarachnoid hemorrhage [CONSCIOUS-1]).Reference Macdonald, Kassell and Mayer 129 Treatment with either 1, 5, or 15 mg/hour of intravenous clazosentan was started within 56 hours of SAH and continued up to 14 days. The primary endpoint, moderate to severe angiographic vasospasm, was significantly reduced in a dose-dependent fashion from 66% in the placebo group to 23% in the 15-mg/hour clazosentan group (risk reduction, 65%; 95% confidence interval, 47% to 78%; p < 0.0001). There was no clear effect on infarction or outcome, and clazosentan was associated with increased rates of pulmonary edema, hypotension, and anemia. CONSCIOUS-2 was a multicenter placebo-controlled phase III study examining the effect of 5 mg/hour clazosentan, given for up to 14 days after aneurysm clipping (n = 768) versus placebo (n = 389).Reference Macdonald, Higashida and Keller 130 The 6-week primary composite endpoint included all-cause mortality, vasospasm-related new cerebral infarcts, delayed ischemic neurological deficit because of vasospasm, and initiation of rescue therapy for vasospasm. The main 12-week secondary endpoint was functional outcome using the extended Glasgow outcome scale. Clazosentan at this dose had no significant effect on mortality, vasospasm-related morbidity, or functional outcome. Lung complications, anemia, and hypotension were more common with clazosentan. Subgroup analysis indicated that clazosentan did reduce vasospasm-related morbidity in poor-grade patients and those with diffuse, thick SAH at baseline, without improving functional outcome in those same groups. CONSCIOUS-3 was a similar phase III trial of clazosentan in patients whose aneurysms were secured by endovascular coiling as opposed to clipping, but in additional to the dose of 5 mg/hour included a 15-mg/hour treatment group.Reference Macdonald, Higashida and Keller 131 This study was halted prematurely after CONSCIOUS-2 results were known. Again, no overall benefit from treatment with clazosentan was seen in terms of functional outcome, although the higher dose did significantly reduce vasospasm-related morbidity/all-cause mortality.
It is unclear why clazosentan, which had been shown to reduce vasospastic arterial narrowing, did not also reduce the incidence of poor outcome, but the authors suggest it could be due to methodological problems, inadequate sample size, insensitivity of clinical outcome measures, imbalance in the use of rescue therapy, or because mechanisms other than vasospasm (and upon which the drug has no effect) also contribute to poor outcome in an important way.
Statins
Statin agents inhibit 3-hydroxyl-3-methylglutaryl coenzyme A reductase, which is the rate-limiting enzyme in the mevalonate pathway of cholesterol synthesis. In addition to their hypolipidemic activity of lowering cholesterol levels, they have other salutary effects on the cardiovascular system, including improving endothelial function, modulating inflammatory responses, maintaining the stability of atherosclerotic plaque, and preventing thrombus formation. Perhaps important as they relate to SAH, statins also improve CBF by upregulation of eNOS. Statins increase NO biosynthesis and bioavailability, which gave them great potential for vasospasm prophylaxis in the setting of SAH.Reference O’Driscoll, Green and Taylor 132
In a pilot randomized clinical trial, simvastatin (80 mg) given once daily for 14 days appeared to decrease the incidence of radiographic vasospasm and delayed ischemia.Reference Lynch, Wang and McGirt 133 Reduced vasospasm and cerebral infarction were also found in several cohort studies comparing SAH outcomes in statin users and nonusers at the time of hospital admission.Reference Parra, Kreiter and Williams 134 , Reference McGirt, Blessing and Alexander 135 A phase II placebo-controlled trial of 80 patients randomized to receive either placebo or 40 mg of oral pravastatin for up to 14 days found that statin use reduced vasospasm and severe vasospasm by 32% and 42%, respectively, and reduced vasospastic infarcts and mortality by 83% and 75%, respectively (all significant p values).Reference Tseng, Czosnyka, Richards, Pickard and Kirkpatrick 136 The improvement in early outcome proved durable at 6 months.Reference Tseng, Hutchinson, Czosnyka, Richards, Pickard and Kirkpatrick 137 A small single-center randomized controlled trial of simvastatin 80 mg once daily versus placebo found a significant reduction in cholesterol and low-density lipoprotein in the statin-treated patients, but not in any other parameter including delayed cerebral ischemia and poor outcome.Reference Vergouwen, Meijers and Geskus 138 Another cohort study indicated that although the cholesterol-lowering agent atorvastatin reduced the incidence of vasospastic infarction seen on CT scanning, it did not improve clinical outcome.Reference Sanchez-Pena, Nouet and Clarencon 139 A meta-analysis of six randomized controlled statin trials found that, although delayed ischemic deficits were less common in the statin-treated patients, there was no overall significant reduction in poor neurological outcome.Reference Su, Xu, Hai, Wu and Yu 140 The international, randomized, and double-blind trial Simvastatin in Aneurysmal Subarachnoid Hemorrhage, which recruited 803 patients until February 2013, failed to detect any benefit in the use of simvastatin, 40 mg per day, started within 96 hours of ictus then for up to 3 weeks, for either short- or long-term outcomes.Reference Kirkpatrick, Turner, Smith, Hutchinson and Murray 141 High-dose simvastatin (80 mg per day for 3 weeks) was compared with 40 mg per day for 3 weeks in a randomized, controlled, double-blinded clinical trial that recruited 255 patients.Reference Wong, Chan and Siu 142 No difference was seen between the high- and low-dose groups in the incidence of delayed ischemic deficits or clinical outcome. At present, it would appear that patients with SAH do not benefit from the addition of statins during the acute stage.
Tirilazad Mesylate
Tirilazad, an inhibitor of iron-dependent lipid peroxidation and free radical scavenger, was tested in a series of large-scale randomized, controlled trials and became licensed for use in several countries, although not in North America. An analysis of all tirilazad results showed decreased mortality associated with treatment, but other good-outcome endpoints did not show benefit except in a post hoc analysis of poor-grade patients.Reference Dorsch, Kassell and Sinkula 143 Because of gender-specific pharmacokinetics, higher dosages were necessary in women, who appeared to benefit less than men.
Reversal of Vasospasm and Cerebral Ischemia: Rescue Treatment
Augmentation of CBF through and via collaterals around vasospastic vessels by elevating systemic blood volume and pressure can reverse cerebral ischemia, and either pharmacologic or balloon dilation of narrowed arteries can reverse vasospasm itself. When and how these two “reversal” or “rescue” treatments should be implemented and combined is subject to some variation.
Triple-H Therapy: Hypervolemia, Hypertension, and Hemodilution
This combination is intended to improve cardiac output, increase CPP, and optimize hemorheology for oxygen transport. A degree of hemodilution accompanies any deliberate volume expansion, and reduced viscosity may contribute to an improvement in oxygen delivery, provided that the hematocrit (i.e. the oxygen-carrying capacity) does not fall below 30 and the hemoglobin concentration is maintained higher than 9 g/dl.
When symptomatic vasospasm is diagnosed or strongly suspected, hemodynamic treatment can commence by additional volume expansion with an isotonic crystalloid infusion, which in euvolemic patients raises CBF in vasospastic territories without a significant change in cardiac indices.Reference Jost, Diringer and Zazulia 144 A replete intravascular volume beyond which additional expansion is probably of no further benefit corresponds to a central venous pressure between 8 and 10 mmHg or a pulmonary capillary wedge pressure in the range of 14 to 16 mmHg. Unless patients have a complicated cardiopulmonary status (because of myocardial infarction, heart failure, or pulmonary edema, for example), a pulmonary artery catheter is unnecessary.Reference Shure 145 , Reference Mutoh, Kazumata and Ajiki 146 There is evidence that aggressive hemodynamic monitoring with a transpulmonary thermodilution monitoring system (PiCCO Plus, Pulsion Medical Systems SE, Feldkirchen, Germany) that measures cardiac output, global end-diastolic volume index, extravascular lung water index, pulmonary vascular permeability index, and systemic vascular resistance index, and then administration of fluid therapy directed by these parameters to optimize hemodynamic status, is beneficial in poor-grade patients.Reference Yoneda, Nakamura and Shirao 147 “Early goal-directed fluid therapy” using PiCCO monitoring was associated with significantly less delayed cerebral ischemia and better functional outcome compared with standard fluid therapy (guided by fluid balance and a central venous catheter), although the difference was only seen in poor-grade patients.Reference Mutoh, Kazumata, Terasaka, Taki, Suzuki and Ishikawa 148 Induced hypertension is more effective than aggressive hypervolemia in improving cerebral oxygenation in patients with vasospasm and has fewer complications.Reference Raabe, Beck, Keller, Vatter, Zimmermann and Seifert 149 Provided that the ruptured aneurysm has been repaired, symptomatic vasospasm should be treated by the administration of a vasopressor, the most commonly used being phenylephrine or norepinephrine.Reference Meyer, Deem, Yanez, Souter, Lam and Treggiari 150 The more purely α-agonist vasopressors, norepinephrine (titrated to a maximum dose of 20 μg/kg per minute) and phenylephrine (titrated to a maximum dose of 180 μg/kg per minute), or a combination of the two, should be commenced promptly. Dobutamine or dopamine can be considered, which at low to moderate doses have mainly β-agonist, inotropic effects. Another cardiac inotrope, milrinone, has also been used after SAH.Reference Naidech, Du and Kreiter 151 Blood pressure rises with elevations in cardiac output, which increases CPP. However, if a prompt blood pressure response does not occur with an inotrope (i.e. to dopamine administered at 10 to 15 μg/kg per minute), either epinephrine or norepinephrine should be added. At high doses, dopamine also causes α-adrenergic agonism; however, it is accompanied by unwanted tachycardia because of β2 receptor activity, which norepinephrine does not possess.
The key aspect of treatment is rapid elevation of blood pressure, regardless of the agent chosen. Systolic blood pressure of 200 mmHg or higher or CPP greater than 80 mmHg is sometimes required, but if ischemic signs persist at systolic pressures in this range or higher, hypertensive treatment should be considered to have failed. Blood pressure elevations this extreme might also aggravate cerebral ischemic cerebral edema leading to dangerous elevations in intracranial pressure.
It should be noted that hypertension and hypervolemia do not seem to increase the risk for hemorrhage from unsecured, unruptured aneurysms in the acute setting or in their short-term natural history.Reference Hoh, Carter and Ogilvy 152
The significant risks of triple-H therapy are cardiac failure and infarction, pulmonary edema, the complications associated with either central venous catheter or pulmonary artery catheter placement, and possibly cerebral edema and raised intracranial pressure.Reference Rosenwasser, Jallo, Getch and Liebman 153 - Reference Amin-Hanjani, Schwartz, Sathi and Stieg 155 Risks are greatest in the elderly and in patients with intrinsic, preexisting cardiopulmonary disease.
Intra-aortic Balloon Counterpulsation
Originally designed for the management of cardiogenic shock, placement of a transfemoral aortic balloon that inflates on aortic valve closure with each cardiac cycle and augments diastolic flow proximally to the coronary and cranial arteries and distally to the peripheral circulation has been reported to be feasible and effective in patients with combined severe vasospasm and cardiac failure.Reference Apostolides, Greene, Zambramski, Fitzgerald and Spetzler 156 , Reference Nussbaum, Sebring, Ganz and Madison 157 Administration of intra-aortic balloon counterpulsation as vasospasm prophylaxis for patients at high risk of vasospasm but normal cardiac function did not confer any clinical benefit in a small, single-center randomized controlled trial.Reference Bulters, Birch and Hickey 158
Endovascular Reversal of Vasospasm
If the hemodynamic goals of triple-H therapy are not easily met in a patient with persistent signs of cerebral ischemia, if the signs do not reverse within several hours of hypertension induced into the target range, or if a patient has a fragile cardiopulmonary status, it is appropriate to proceed directly to endovascular treatment of vasospasm. It is important to first rule out a large cerebral infarct with CT because dilation therapy is both futile and dangerous in this situation.
In the setting of symptomatic vasospasm, angiography will show which segments of the intracranial vasculature are affected, but this must be correlated with clinical examination and TCD results. The internal carotid and basilar arteries can usually be safely dilated using currently available endovascular balloons, as can the first (M1) segment of the MCA. Mechanical balloon angioplasty is preferably performed within 2 hours of symptom onset for best results.Reference Rosenwasser, Armonda, Thomas, Benitez, Gannon and Harrop 159 Balloon catheters are selected to match the baseline, normal diameter of the arterial segment in vasospasm. Choosing an oversized balloon can result in catastrophic vessel rupture, so it is important to examine the patient’s prior, prevasospastic imaging, and in part to ensure that a hypoplastic artery is not mistaken for vasospasm. Once the balloon is selected, the wire and then balloon are navigated across the site of stenosis. When the narrowed segment is long, it is sometimes simplest to place the balloon distally; following balloon inflation, the balloon is deflated, pulled proximally, and reinflated until the entire segment has been dilated.
The efficacy of mechanical balloon angioplasty was nearly 100% in a recent series, and associated with a complication rate of only 1%.Reference Chalouhi, Tjoumakaris and Thakkar 160 These reported results are significantly better than older series, where efficacy ranged from 11 to 93%, associated clinical improvement rate of 328/530 (62%), and a major complication rate of 5%, including vessel rupture in 1.1%.Reference Hoh and Ogilvy 161
Canine models of vasospasm have shown that the results of dilation are durable for up to 3 weeks.Reference Megyesi, Vollrath, Cook, Chen and Findlay 162 , Reference Megyesi, Findlay, Vollrath, Cook and Chen 163 This mirrors clinical experience, in which balloon angioplasty is typically effective for the remainder of the vasospasm episode and patients rarely require repeat balloon dilation.Reference Grande, Nichols and Khan 164 - Reference Elliott, Newell and Lam 166 However, mechanical dilation is only effective over the segment of the artery where the balloon was inflated, and it is possible to develop symptomatic vasospasm later in an adjacent, usually distal arterial segment. Although very uncommon, reintervention can occasionally be required for balloon dilation of the same vasospastic segmentReference Frontera, Gowda and Grilo 167 ; much more commonly, repeat treatment is required for adjacent arterial segments.Reference Santillan, Knopman, Zink, Patsalides and Gobin 168 , Reference Tekle, Chaudry and Hassan 169
Although balloon angioplasty is usually reserved for larger (≥2.0 mm diameter) vessels, there is one recent report of 175 safe angioplasties in distal vessels without procedure-related symptomatic complications.Reference Santillan, Knopman, Zink, Patsalides and Gobin 168 Those authors recommend using a calibrated balloon to decrease the risk of vessel rupture.
Further technical pitfalls to avoid include balloon inflation next to a fresh coil mass or recently placed clips because the expansion of the balloon may disrupt the coils or clips, with potential hemorrhagic or ischemic consequences.
For vasospasm affecting more distal vessels, where the operator judges that the risks of angioplasty are too great, local delivery of vasodilators might help establish sufficient vessel caliber to prevent infarction. The use of local intra-arterial milrinone in this setting is currently of interest.Reference Fraticelli, Cholley, Losser, Saint Maurice and Payen 170 , Reference Shankar, dos Santos, Deus-Silva and Lum 171 Milrinone, a selective phosphodiesterase III inhibitor, can be administered through a simple microcatheter or a deflated dual-lumen balloon catheter placed proximal to the spastic territory. Hemodynamic instability can be encountered, so slow infusions (over 30-40 minutes) are preferable, with close attention paid to blood pressure and heart rate. Other intra-arterial vasodilators such as papaverine, nifedipine, verapamil, and nimodipine have been also used for vasospasm in a similar manner, with inconsistent results. Most important, in our opinion, is that when distal vasospasm beyond the reach of the balloon catheter is seen at the end of the intervention it is important to maintain triple-H treatment afterwards and in particular induced hypertension, until the vasospasm period is over (Figures 3 and 4).
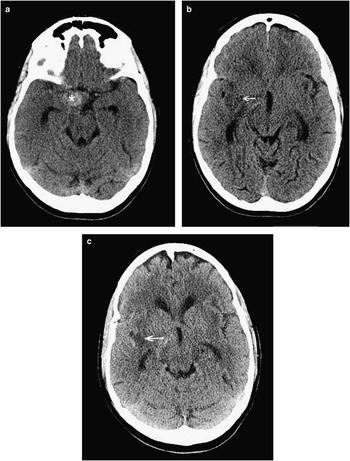
Figure 3 This 56-year-old woman was diagnosed with subarachnoid hemorrhage 10 days following her thunderclap headache, and CT was finally done for persistent headache and new-onset left-sided numbness and weakness. CT scanning showed a spherical hyperdensity in the right basal cisterns (A, asterisk), which was a large aneurysm as well as ischemic hypodensities in the left frontotemporal peri-insular brain tissue (B, C; arrows). Ten days following rupture, the subarachnoid blood had largely cleared from the subarachnoid cisterns (see Figure 4).
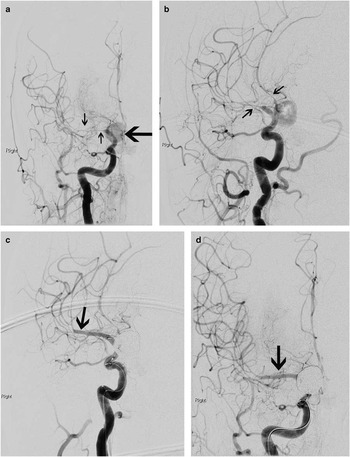
Figure 4 By the time of catheter angiography several hours later, the patient’s entire left side had become paralyzed. Angiography showed a large, sluggishly opacifying, right-sided posterior communicating artery aneurysm (large arrow, A), critical right middle cerebral artery vasospasm, and narrowing of the anterior cerebral arteries as well (A, B; small arrows). The aneurysm was occluded with coils and then the M1 segment of the middle cerebral artery underwent balloon angioplasty (C, D; arrows) followed by induced hypertension for persistent vasospasm of the more distal middle cerebral as well as anterior cerebral arteries. She recovered spontaneous movement on her left side promptly, and follow-up CT scanning showed no extension of her infarction. She went on to make a good recovery over 6 months.
Intravenous Milrinone and Other Reversal Therapies
Milrinone is a selective phosphodiesterase III inhibitor affecting cyclic adenosine monophosphate pathways, with both inotropic and vasodilatory properties.Reference Vroom, Pfaffendorf, van Wezel and van Zwieten 172 There have been scattered reports of milrinone’s potential positive effect on both experimental and clinical vasospasm when administered either into the subarachnoid cisterns, intravenously, or intra-arterially.Reference Fraticelli, Cholley, Losser, Saint Maurice and Payen 170 , Reference Nishiguchi, Ono, Iseda, Manabe, Hishikawa and Date 173 - Reference Arakawa, Kikuta, Hojo, Goto, Ishii and Yamagata 175 A retrospective case series consisting of 88 patients with symptomatic vasospasm treated with intravenous milrinone between 1999 and 2006 has been reported. Reference Lannes, Teitelbaum, del Pilar Cortés, Cardoso and Angle 176 The “Montreal Neurological Hospital protocol” consists of a milrinone 0.1-0.2 mg/kg intravenous bolus followed by a 0.75 mcg/kg/minute infusion, combined with optimal hydration and correction of any electrolyte or glucose abnormalities. If the patient failed to improve after 30 minutes, the infusion was increased to 1.25 mcg/kg/minute, and vasopressor infusions were begun to either restore or achieve blood pressure targets when necessary (68% of patients). Milrinone infusions were continued for a mean of 10 days and were considered by the authors as safe. Only one patient received intra-arterial milrinone and no angioplasties were carried out in this large case series. Roughly 40% of patients demonstrated vasospasm-related infarcts on CT scanning. This experimental treatment strategy with intravenous milrinone has not yet been reported in a controlled study, and it is premature at this time to consider it a vasospasm treatment “protocol.”
In patients with severe and medically refractory vasospasm, sodium nitroprusside, an NO donor, has been administered into the lateral ventricles, either as single injections or as a continuous infusion via a ventriculostomy catheter, but the results have not been consistent or promising.Reference Thomas and McGinnis 177 , Reference Raabe, Zimmerman, Setzer, Vatter, Berkefeld and Seifert 178 Cervical sympathetic blockade with regional anesthesia appeared to result in improvement of mild to moderate symptoms of vasospasm, but without reversal of angiographic vasospasm.Reference Treggiari, Romand, Martin, Reverdin, Rufenacht and De Tribolet 179
A Practical Approach to the Prevention and Treatment of Vasospasm
Patients should be kept well-hydrated with isotonic crystalloid (at least 3 L/day, combined), intracranial pressure is kept normal with the liberal use of external ventricular drainage, and CPP is optimized to levels higher than 70 mmHg. No attempt need be made to control mild or moderate hypertension (Table 7). Daily TCD examinations are valuable to screen for developing vasospasm, and values in excess of 200 cm/second in the MCA are indicative of significant angiographic vasospasm in that vessel. Regular clinical assessment to search for subtle changes in mentation, verbal output, and arm and hand control is critical. Obtunded or sedated patients being ventilated should be considered for direct vascular imaging (computed tomographic angiography is useful except for examination of the arteries immediately adjacent to the clip[s] or coils) on post-SAH day 5 and then again several days later if considered necessary.
Table 7 Vasospasm management

All patients should receive nimodipine, 60 mg every 4 hours by mouth or via a nasogastric tube for 21 days or as long as they remain in the hospital, phenytoin should be avoided unless the patient has a documented seizure, and corticosteroids are not recommended. Every effort should be made to avoid hyponatremia, fever, and hypoxia, and patients should be fed as soon as possible.
Symptomatic vasospasm and moderate to severe angiographic vasospasm in comatose patients should be treated by infusion of a vasopressor. For most patients, central venous catheters are preferable to pulmonary artery catheters, and euvolemia or slight hypervolemia should be ensured. If signs do not respond quickly and completely or a target blood pressure and CPP are difficult to reach, it is recommended to move directly to endovascular treatment with angioplasty for all reachable, symptomatic segments of vasospasm. A CT scan should be performed first to rule out a large, established infarction or a new hemorrhage. In general, one should not wait until medical treatment of symptomatic vasospasm has utterly failed over a period of hours before initiating endovascular treatment, because under these circumstances angioplasty is more often inconvenient, dangerous, and unsuccessful.
Conclusion
The most important testing for developing vasospasm and cerebral ischemia remains regular bedside neurological examinations by an experienced nurse or physician for the first two weeks following a Fisher grade 2 or higher SAH (Table 1). Transcranial Doppler is a helpful addition, bearing in mind the factors other than vasospasm that cause blood velocities to increase, making false-positive findings a hazard. Continuous monitoring of brain tissue oximetry or blood flow with a parenchymal probe or scalp patch share the drawback of surveying only a small volume of brain tissue in a single arterial territory (false-negative results therefore being a concern). CTP provides a useful and informative picture of the whole brain, but is a “snap shot in time,” and like vascular imaging itself needs to be timed carefully to maximize utility (Table 5).
It is disappointing to report that recent large, randomized controlled trials did not demonstrate that the endothelin antagonist clazosentan, the cholesterol-lowering agent simvastatin, and the vasodilator magnesium sulfate prevent vasospasm, reduce delayed ischemia, or improve patient outcome following SAH. Those trials are reminders of the critical role of randomized trials in proving efficacy because all of these agents showed very promising results in preliminary studies.
The pathophysiology of vasospasm following SAH is complex and up until now has defied any simple pharmacological solution. The single and partial exception is the calcium antagonist agent nimodipine, the only treatment for cerebral vasospasm and delayed ischemia that presently achieves a class I, level of evidence “A” recommendation in the American Heart and Stroke Association aneurysmal SAH management guidelines.Reference Connolly, Rabinstein and Carhuapoma 180 Nimodipine has been in use for nearly 30 years, but still has an uncertain mechanism of action and an overall only modest beneficial effect on outcome.
Reduction in vasospasm-related morbidity and mortality in the past two decades has been in a greater part because of appropriate fluid management following SAH, induced hypertension when indicated for symptomatic ischemia, and judicious use of balloon angioplasty (Table 7), although these treatments will never be validated in a placebo-controlled randomized trial.
Acknowledgments
The authors thank Ms. Shelan Beatty for expert assistance in preparing this review and dedicate it to our neuro- intensive care nurses who do all the hard work in looking after our patients with aneurysmal subarachnoid hemorrhage, 24/7.
Disclosure information :
The authors do not have anything to disclose.












 Open access
Open access