


Case Presentation: Dr. Mandzia
A 68-year-old right-handed male initially presented with a 3-week history of dizziness and blurred vision with unsteadiness on his feet. The dizziness was described as a sensation of the ground moving. No rotational component was described. He had no nausea, vomiting, fever, diplopia, hearing loss, headaches, or preceding viral illness.
His medical history was significant for paroxysmal atrial fibrillation, hypertension, hyperlipidemia, remote L5-S1 discectomy, chronic low back pain, chronic bilateral tinnitus (presumed as resulting from occupational noise exposure), anosmia following a remote head injury, and microscopic hematuria (with a negative workup).
His medications were escitalopram 20 mg every night at bedtime, amiodarone 200 mg once daily (OD), metoprolol, 50 mg twice daily, hydrochlorothiazide 12.5 mg OD, acetylsalicylic acid 81 OD, and rabeprazole 20 mg OD and acetaminophen with codeine (300 mg and 30 mg, respectively) as needed. He had no known drug allergies.
He had a 27 pack-year smoking history. He denied any alcohol intake in the preceding 18 months. He was a retired army/corrections officer, married with three children. His family history was notable for ischemic heart disease.
His general examination was normal and his neurological exam was only noteworthy for his unsteadiness. Head CT scans were negative. He was diagnosed with labyrinthitis and treated with prednisone 30 mg×5 days and betahistine. His symptoms improved and he was discharged home after 5 days to follow-up with the departments of ear, nose, and throat, and ophthalmology.
One week after discharge, he began to experience progressive, bilateral lower extremity numbness and weakness, with difficulties in ambulation requiring a walker. He had experienced urinary retention and incontinence and fecal incontinence on one occasion. He noted reduced perianal sensation.
The patient was afebrile, with a blood pressure of 126/78 and a heart rate of 68 per minute. The neurological examination was normal for cranial nerves and upper extremities. However, the lower extremity examination was notable for bilateral leg spasticity right > left, mild proximal weakness (4– to 4+/5), increased reflexes in the knees (3), sustained clonus in the right ankle, and an absent left ankle reflex. Sensory examination revealed a T10 sensory level to pin prick, absent vibration to the right tibial tuberosity, and decreased vibration at the left great toe. His gait was unsteady and he was unable to do a tandem walk. There was no finger to nose ataxia. He had decreased rectal tone and perianal sensation.
Initial investigations revealed a normal hemoglobin and white blood cell count, but there was thrombocytopenia (range, 41-86). The mean corpuscular volume was 97.7 fL (normal range, 80-95). Vitamin B12 levels were 97 pmol/L (normal, >156). The erythrocyte sedimentation rate (ESR) was 33 and C-reactive protein (CRP) 24.9. His postvoid residual urine volume was 400 ml.
A lumbar puncture was performed and cerebrospinal fluid (CSF) analysis revealed a normal glucose of 3 mmol/L (normal, 2.2-3.9), an elevated protein of 513 mg/L (normal range, 200-400) with cytology revealing eight nucleated cells and 126 red cells in tube 1, compared with four nucleated cells and no red cells in tube 4 (81% of nucleated cells were lymphocytes). CSF culture, cytology, oligoclonal bands, angiotensin-converting enzyme levels, and Venereal Disease Research Laboratory (VDRL) screens were negative.
Antiphospholipid antibodies and lupus anticoagulants were negative. HIV serology, Lyme serology, VDRL, and hepatitis B and C surface antibodies were negative. Hepatitis B core antibodies were present.
Discussion: Dr. Watling
The patient’s initial symptoms of dizziness and blurred vision are nonspecific. His description of the dizziness as a feeling of the ground moving suggests vertigo, but I do not think there is enough information to allow localization of this vertigo as either central or peripheral.
The subsequent presentation of progressive lower extremity weakness and numbness is more readily localizable to the mid- to lower thoracic spinal cord. The bilateral and rather symmetric weakness of the legs with upper motor neuron signs, along with urinary and fecal incontinence, reduced perianal sensation, and a T10 sensory level, strongly support this localization. The only sign that is an outlier is the absent left ankle jerk, although the history of back pain and prior disc disease at L5-S1 raises the possibility that this sign is old and unrelated to the presenting illness. One symptom notable by its absence is new or increasing back pain, which is typically a feature of compressive lesions of the spinal cord.
The laboratory investigations are notable for a reduced (and falling) platelet count and modest elevations of CRP and ESR. Similarly, the CSF is abnormal, with a minor lymphocytic pleocytosis and modest elevation of protein, but these results are nonspecific.
The differential diagnosis is that of an acute to subacute thoracic myelopathy, and includes the following.
-
1. Compressive lesions, including metastatic or primary tumours of the spine. Against this possibility is the absence of new or increasing back pain, which would be unusual in cases of epidural spinal cord compression.
-
2. Inflammatory lesions of the spinal cord, such as transverse myelitis. Collagen vascular diseases and sarcoidosis also merit consideration as potential causes of inflammatory myelopathies, although the laboratory results provided do not lend support to either of these possibilities.
-
3. Vascular lesions of the spinal cord. Infarction and hemorrhage are unlikely with the progressive nature of the symptoms, but vascular malformations, including a dural arteriovenous fistula, or vasculitis of the spinal cord might present in this manner.
-
4. Unusual malignancy-related causes, including intravascular lymphoma or leptomeningeal carcinomatosis or lymphomatosis.
The B12 deficiency merits correction, given its potential neurological consequences, but it is unlikely that B12 deficiency is a sufficient explanation for such an acute presentation. I have some difficulty linking the thrombocytopenia to the presentation; although thrombocytopenia can occur in lupus or in the antiphospholipid antibody syndrome, we have no serologic support for these diagnoses.
Although it is difficult to link the initial vertigo with the subsequent myelopathy, the chronological proximity of these two events suggests that they may be related. The localizations of these presentations are clearly different, so linking them would require a diagnosis capable of producing multifocal nervous system dysfunction with a relapsing and remitting course. Possibilities would include multiple sclerosis (but the patient’s age would make a new diagnosis of multiple sclerosis unlikely), sarcoidosis, collagen vascular disease, antiphospholipid antibody syndrome, or vasculitis.
The key initial investigation would be an MRI of the spine, because compressive lesions would require urgent treatment and must not be missed. Depending on the results of this MRI, other useful investigations might include an MRI of the brain with magnetic resonance or CT angiography and a repeat CSF analysis.
Course in The Hospital: Dr. Mandzia
The patient was treated for transverse myelitis with a 5-day course of intravenous methylprednisolone. There was partial improvement of urinary continence and leg strength (grade 5 with the exception of hip flexors, which were grade 4), and he could ambulate short distances (300 m) with a walker. His unsteadiness and sensory loss remained. His low platelets were ascribed to idiopathic thrombocytopenic purpura by the hematology department. He was transferred to rehabilitation.
Two days later, there was evidence of declining leg strength and mobility, making transfers and ambulation unmanageable. He also complained of intermittent horizontal diplopia and a painful sensation across his abdomen.
On examination, his cranial nerves were normal. Tone remained increased in the lower extremities, but strength had diminished to 3– to 4– bilaterally. Reflexes were graded 2 throughout and plantar reflexes were downgoing. He was found to have allodynia at T10, an unchanged T10 sensory level to pin prick, and absent vibration sense to the knees.
His nerve conduction studies showed evidence for a left L5 radiculopathy and confirmed no evidence of peripheral neuropathy or features of Guillain-Barre syndrome.
He received a 3-day course of 500 mg intravenous methylprednisolone followed by 2 g/kg intravenous immunoglobulin over 2 days. His power testing fluctuated, but in general he had some clinical improvement of his motor strength and was discharged back to rehabilitation.
His condition continued to show modest improvement under rehabilitation until he began to deteriorate again with progressive leg weakness, horizontal diplopia, memory difficulties, depression, and fatigue over the course of several days. Investigations revealed continued elevations of CRP (75) and ESR (50), thrombocytopenia (75), and anemia (hemoglobin 96).
When initially examined, he was alert and oriented and could follow two-step commands, but exhibited difficulty naming of low-frequency items. He had a right esophoria, decreased right lateral gaze with left-beating nystagmus on left lateral gaze. Lower extremity power was absent except for grade 2 and 3 strength of right and left hip flexion, respectively. The right lower extremity remained spastic. He had brisk reflexes, graded 3 throughout with bilateral crossed adductors, ankle clonus on the right and upgoing plantar reflexes. A pin prick sensory level at T10 remained, and vibration was now absent up to the anterior superior iliac spines.
His investigation included a repeat lumbar puncture (protein, 781; glucose, 3.1; 15 and 7 nucleated cells; 71% lymphocytes; negative culture and cytology; positive immunoglobulin [Ig]G index), and a negative vasculitic screen (negative antinuclear antibodies, double-stranded DNA, complement, anti-extractable nuclear antigens, perinuclear antineutrophil cytoplasmic antibodies [p-ANCA], and cytoplasmic antineutrophil cytoplasmic antibodies [c-ANCA]). Rheumatoid factor was elevated at 27 IU/ml (normal, <21). CT scans of thorax, abdomen, and pelvis were unremarkable apart from splenomegaly.
Imaging: Drs. Bahakeem and Leung
The initial CT and MRI of the brain and spine demonstrated no acute changes. At L4-5 and L5-S1, chronic degenerative spondylotic changes and distant postoperative changes were present. There was encroachment on the left L5 root, but no cord compression or signal change.
Two months later, MRI brain revealed several small foci of diffusion high signal in the pons, left posterior limb of internal capsule, adjacent cerebral peduncle, deep gray structures, and posterior cerebral hemispheres bilaterally (Figure 1A). There was a corresponding increased signal on T2-weighted imaging (T2WI) and fluid-attenuated inversion recovery (FLAIR), but no significant mass effect, hemorrhage, or enhancement (Figure 1B). MRI of the spine demonstrated patchy small areas of increased signal on T2WI in the spinal cord dorsally at T10 and T11 (Figure 2A, B). The intracranial findings were compatible with small infarcts in multiple vascular territories from a central embolic source, hypercoagulable state, vasculitis, or rarely a consequence of intravascular lymphoma. These diagnostic considerations could account for the spine findings as well, although inflammatory and infectious etiologies were also within the differential.
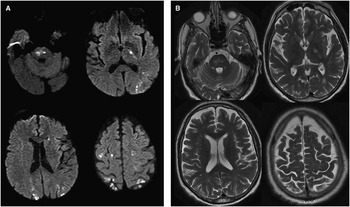
Figure 1 (A) Axial diffusion-weighted imaging (DWI) demonstrates multiple restricted diffusion signal intensities in the pons, posterior limb of left internal capsule, deep gray matter, and bilateral posterior cortices. (B) Axial T2WI reveals high signal intensities without mass effect that colocalize with the restricted DWI abnormalities.
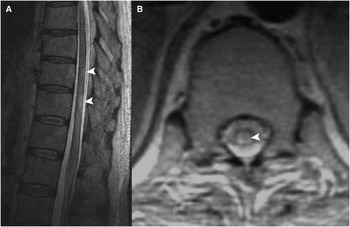
Figure 2 (A) Sagittal T2WI demonstrates patchy increased signal intensity at the T10-T11 level. (B) axial T1-weighted imaging demonstrates a corresponding dorsal intramedullary lesion.
Cerebral angiography demonstrated multiple areas of irregular distal small vessel stenosis with a poor parenchymal blush in the anterior cerebral artery and middle cerebral artery territories bilaterally (Figure 3). The proximal branches of the circle of Willis were normal in caliber. The posterior circulation and venous drainage were unremarkable. With the MRI findings, the small vessel stenoses were compatible with vasculitis.

Figure 3 Cerebral angiography (coronal view) with selective injection of the left internal carotid artery demonstrates irregularities in small vessels in the distal left anterior and middle cerebral distributions.
Final Discussion: Dr. Watling
Neurology is surprising and humbling. As this patient’s illness evolved, the picture of a multifocal central nervous system (CNS) process became clearer. In addition to clinical evidence of a progressive myelopathy, the developing cognitive and language dysfunction suggests involvement of the cerebral cortex, whereas the nystagmus and diplopia implicate the brainstem. Multifocal ischemic lesions could result from a number of processes, including underlying cardiac pathology leading to repeated emboli or hypercoagulable states. In this case, however, the imaging shows blood vessel abnormalities compatible with vasculitis, and repeat CSF analysis shows persistent, nonspecific abnormalities compatible with an inflammatory process. With no clinical or serologic evidence of systemic vasculitis, the most likely diagnosis is primary CNS vasculitis. This entity had not figured prominently in my initial differential diagnosis, primarily because of the initial presentation with myelopathy, which is distinctly unusual in CNS vasculitis. The important lesson here may be to attend carefully to the history, which suggested from the start that the symptoms of myelopathy were actually part of a multifocal, fluctuating process.
Biopsy is challenging in cases of suspected CNS vasculitis; choosing a target for tissue sampling is difficult, and findings may be nondiagnostic. In this situation, I would treat with corticosteroids and cyclophosphamide based on the information available, and would not feel that a biopsy would be likely to enhance the therapeutic decision-making.
Treatment and Course: Dr. Mandzia
The patient was diagnosed with primary angiitis of the CNS and he received pulse methylprednisolone, 1 g for 3 days, then prednisone 50 mg daily as well as monthly intravenous cyclophosphamide. Further workup excluded cardioembolic sources.
He developed delirium and achieved no substantive neurological improvement despite continued treatment. He was transferred back to his regional hospital. He had further confusion and delusion and his prednisone dosage was decreased to 20 mg. Over the following week, he had declined further prompting palliative measures and the patient expired 2 weeks thereafter.
Pathology: Dr. Alturkustani
A complete postmortem examination including the brain and spinal cord was performed. The general autopsy showed no evidence of systemic inflammatory disease. The fresh brain weight was 1307 g and appeared grossly intact externally on coronal sections of the cerebral hemispheres and axial sections of brainstem and cerebellum.
Microscopic examination revealed multiple tiny infarcts in the cerebral cortex, white matter (Figure 4A, B), and deep gray structures, including basal forebrain, hypothalamus, and internal capsule.

Figure 4 Photomicrographs of cerebral and spinal cord pathology (hematoxylin and eosin). (A) Multiple small infarcts in the cerebral cortex (arrowheads). (B) Subcortical white matter rarefaction. (C) Spinal cord infarcts predominantly affecting the gray matter and posterior columns. (D) Spinal cord parenchymal vessel with intramural lymphocytes.
Most microinfarcts were subacute to chronic with small cavities filled with macrophages. There were no acute infarcts, acute inflammation, microorganisms, or inclusions. A few small leptomeningeal arteries, cerebral arterioles, and venules had light perivascular lymphocytic infiltrates. Such infiltrates were more prominent around small arteries of the spinal cord (Figure 4C, D) and were predominantly composed of T cells expressing CD3 and CD8, with negligible CD4-positive T and B cells (CD20). There was no granulomatous reaction and no vascular amyloid deposition was detected with Congo red stains. Incidental findings included Lewy bodies in the substantia nigra, locus ceruleus, and cerebral cortex.
The principal neuropathological diagnosis was primary CNS vasculitis, with microinfarcts in brain and spinal cord.
Discussion: Dr. Alturkustani
Primary angiitis of the CNS (PACNS) is also known as primary CNS vasculitis, Isolated CNS vasculitis, and granulomatous angiitis of the nervous system. It is defined as vasculitis that is restricted to the brain and spinal cord in the absence of evident systemic inflammatory disease, associated infection, neoplasm, or drug exposure.
PACNS typically affects middle-aged individuals with a peak incidence near age 50. Males are more commonly affected than females (2:1). Most patients present indolently with nonspecific progressive symptoms such as headaches and cognitive dysfunction. However, acute presentation such as strokes (40%), transient ischemic attacks (30% to 50%), aphasia (28%), and seizure (<25%) are not uncommon.Reference Birnbaum and Hellmann 1 Constitutional symptoms present in fewer than 20% of cases and should raise the likelihood of systemic illnesses. Other uncommon presentations include intracranial hemorrhage, symptoms from mass effect, and transverse myelitis.Reference Birnbaum and Hellmann 1
Spinal cord involvement in PACNS was reported in five of 101 patients (5%) in one large review.Reference Salvarani, Brown, Calamia, Christianson, Huston and Meschia 2 Cord involvement can present before, simultaneously, or after cerebral involvement. However, these cases had no distinguishing clinical or laboratory features in comparison to those without cord involvement.
The diagnostic criteria for PACNS were suggested in 1988 by Calabrese and Mallek as an unexplained neurological deficit after thorough clinical and laboratory evaluation, documentation by cerebral angiography, and/or tissue examination of an arteritic process within the CNS, and no other explanation for the angiographic or pathologic features.Reference Calabrese and Mallek 3 Because this definition of PACNS was found to overlap with other vascular disorders, the angiographic features were modified in 2007 by Salvarani et al to “smooth walled segmental narrowing or dilation, and occlusions affecting multiple cerebral arteries in the absence of proximal vessel changes of atherosclerosis or other recognizable causes.”Reference Salvarani, Brown, Calamia, Christianson, Weigand and Miller 4 Birnbaum and Hellmann recommended biopsy confirmation for definitive diagnosis in 2009.Reference Birnbaum and Hellmann 1
Investigation in suspect cases should include basic laboratory testing including routine blood counts with differential, ESR, and CRP and more specialized assays to rule out other systemic inflammatory disorders. Based on context, testing may include: antinuclear antibodies; rheumatoid factor; antibodies to the Ro/SSA, La/SSB, Sm, and RNP antigens; antibodies to double-stranded DNA; ANCA; serum C3 and C4; serum cryoglobulins; serum and urine protein electrophoresis with immune electrophoresis; and finally quantitative Ig levels (IgG, IgM, IgA). CSF analysis is highly sensitive, revealing abnormalities in 80% to 90% of confirmed cases. Furthermore, the combination of an unremarkable MRI and normal CSF analysis has a high negative predictive value for the diagnosis of CNS vasculitis.Reference Stone, Pomper, Roubenoff, Miller and Hellmann 5 The typical findings in CSF include features of aseptic meningitis, normal glucose, raised protein concentrations, and occasionally oligoclonal bands. Excluding infection is essential.
MRI has very high (90% to 100%) sensitivity. The abnormal features include Infarcts (large artery/branches, or small arteries), nonspecific T2 hyperintensities, and gadolinium leptomeningeal enhancement (8%). Cerebral angiography has low sensitivity and specificity,Reference Tartaglia, Pelz, Burneo and Jenkins 6 with the disadvantage of missing small-caliber vascular disease (<1 mm). The typical diagnostic finding is vascular beading (alternating areas of stenosis and dilatation).Reference Hajj-Ali, Singhal, Benseler, Molloy and Calabrese 7
Pathologically, the vasculitic changes present are not highly specific and would fail to reliably distinguish between infectious processes, connective tissues diseases, and PACNS. Instead, the definitive diagnosis is one borne from clinical, radiological, and pathological correlations.
Pathological evidence of vasculitis depends heavily on the biopsy sampling as a recent review concluded that targeted biopsies were positive in 29/46 patients (63%), whereas all nontargeted biopsies (n=5) were negative.Reference Miller, Salvarani, Hunder, Brown, Parisi and Christianson 8 This mirrors our own experience. The role of the biopsy of course is not restricted to confirm the suspected diagnosis, but also to rule out other diagnostic considerations.
Histologically, three patterns have been recognized as diagnostic of PACNS: (1) granulomatous inflammation (most common) that may be associated with amyloid deposition, (2) pure lymphocytic variant (as in the present case), and (3) an acute necrotizing pattern. In all patterns, only intramural inflammation with damage to the vessel wall (with or without fibrinoid necrosis) was considered diagnostic of vasculitis by Miller et al.Reference Miller, Salvarani, Hunder, Brown, Parisi and Christianson 8 Perivascular inflammation is not vasculitis.
Prospective studies and clinical trials have not been conducted in this entity; hence, guidelines are imprecise and based on retrospective studies.Reference Hajj-Ali 9 Nonetheless, these studies recommend (after excluding infection) immunosuppression with cyclophosphamide and glucocorticoids.Reference Hajj-Ali 9 Following these recommendations, patients are empirically started on high-dose glucocorticoids 1 mg/kg/day (to a maximum of 80 mg/day) followed by a tapering course. The duration of cyclophosphamide administration ranges from 3 to 6 months.
Historically, this diagnosis carried a poor prognosis with death following the diagnosis by a mean of 45 days.Reference Sigal 10 With aggressive contemporary immunosuppression, this prognosis has improved with “good outcomes when patients are treated with glucocorticoids or glucocorticoids and cyclophosphamide.”Reference Hajj-Ali 9
D isclosures
None.




