


Many epidemiological studies have revealed that small size at birth or in infancy is associated with adverse health outcomes in adulthood, including diabetes, hypertension and death from IHD(Reference Barker, Bull and Osmond1–Reference Hales, Barker and Clark3). On the basis of these observations, the ‘thrifty phenotype’ hypothesis has been suggested: poor nutritional conditions in early life programme a phenotype in later life in a way that is beneficial to survival under poor nutritional conditions but detrimental when nutrition is abundant(Reference Hales and Barker4). The intra-uterine environment affects fetal growth and development, and brings long-lasting changes to fetal glucose and insulin metabolism. Experimental animal studies have shown that protein restriction in fetal and perinatal life induces a lower growth rate, a reduction in insulin secretion and the loss of glucose tolerance in rats(Reference Hales, Desai and Ozanne5). The size of the pancreatic islets and islet cell proliferation were reduced(Reference Snoeck, Remacle and Reusens6, Reference Cherif, Reusens and Dahri7), and β-cell death by apoptosis was enhanced(Reference Petrik, Reusens and Arany8, Reference Merezak, Hardikar and Yajnik9). These islets were also less vascular(Reference Snoeck, Remacle and Reusens6), and their insulin secretion was reduced(Reference Dahri, Snoeck and Reusens-Billen10). However, the molecular mechanisms responsible for these permanent changes (programming) are not known.
Mitochondria are the intracellular organelles that generate energy for cellular processes by producing ATP and, also, have their own DNA (mitochondrial DNA; mtDNA). Mutations in mtDNA cause diabetes by affecting insulin secretion from pancreatic β-cells(Reference Kadowaki, Kadowaki and Mori11, Reference Ballinger, Shoffner and Hedaya12) because ATP is required for insulin secretion. Additionally, a decrease in mtDNA content has been found to be associated with type 2 diabetes and also insulin resistance(Reference Lee, Song and Shin13, Reference Song, Oh and Sung14). Because mtDNA is transmitted exclusively from the mother and is easily influenced by the environment, changes in the mitochondria have been suggested as a link between poor nutrition in early life and the development of type 2 diabetes in later life(Reference Lee15). Previously, we have reported that the offspring of dams fed a low-protein diet during gestation and lactation had decreased mtDNA content in the pancreas accompanied by a decreased pancreatic β-cell mass(Reference Park, Jin and Cho16), and that the mtDNA content and mtDNA-encoded gene expression of the liver and skeletal muscle in these rats were reduced and did not recover until 20 weeks of age even though proper nutrition was supplied after weaning(Reference Park, Kim and Kim17). Although we showed the pathogenic role of mitochondria in the thrifty phenotype, the mechanisms of how malnutrition in early life induces mitochondrial changes lasting until maturity, despite restoration of nutrition, have not yet been identified.
In this low-protein fetal malnutrition model, the amino acid profile is disturbed in the maternal and fetal plasma as well as in the amniotic fluid. The most affected amino acid is taurine(Reference Reusens, Dahri, Snoeck and Cowett18). Taurine is an amino acid that does not participate in protein synthesis, but has a function in cholesterol excretion, as a neurotransmitter and as a potent antioxidant(Reference Huxtable19). In the fetus, the plasma concentration of taurine is 1·5-fold higher than that of maternal blood, and this level is mostly dependent on transport from the maternal blood through the placenta(Reference Sturman, Gargano and Messing20). A reduced activity of placental taurine transporters results in low fetal taurine levels and fetal intra-uterine growth restriction(Reference Norberg, Powell and Jansson21). There are several reports that taurine supplementation of the maternal low-protein diet in rats completely restores islet cell proliferation and apoptosis, islet vascularisations and insulin secretion from the pancreas of the offspring(Reference Boujendar, Reusens and Merezak22–Reference Cherif, Reusens and Ahn24). These findings suggest that a lack of taurine in the fetus could be a key factor mediating the long-lasting changes (programming) in the thrifty phenotype.
We hypothesised that changes in the mitochondria are critical to the development of the thrifty phenotype, and that taurine may mediate these mitochondrial changes. In the present study, we investigated whether protein malnutrition during gestation and lactation alters both mitochondrial ultra-structures and the expression of mtDNA-encoded genes of the islets in adulthood. In addition, we investigated whether taurine supplementation restores these mitochondrial changes.
Experimental methods
Animals and diets
Male and female Wistar rats were obtained from Jungang Animal Research Ltd (Seoul, South Korea) and housed singly in animal facilities maintained in a temperature- (21 ± 2°C) and humidity- (55 %) controlled room on a standard 12 h light–12 h dark cycle, and were allowed free access to food and water. Diets were purchased from Dyets Inc. (Bethlehem, PA, USA). The control and low-protein diets were isoenergetic and only differed in their protein content; their composition has been described previously(Reference Park, Jin and Cho16, Reference Park, Kim and Kim17).
Virgin female Wistar rats, aged 8 weeks, were caged overnight with males in individual cages (thirty-five rats in each cage) and females were checked daily for vaginal plugs. When pregnancy was confirmed, female rats were randomly divided into three groups (ten rats per group). One group of pregnant rats was fed with a control diet (20 % protein, control group), and a second group with an isoenergetic low-protein diet (LP diet; 8 % protein). The third group of pregnant rats was fed the LP diet supplemented with 2·5 % taurine (Sigma-Aldrich Corp., St Louis, MO, USA) in the drinking water (LP-T diet). These diets were maintained through gestation and 3 weeks of lactation. After weaning, the male offspring in each of the three groups received normal Purina rat chow and tap water (twenty rats per group). Only male animals were chosen for the study to avoid potentially confounding hormonal variables. At 20 weeks of age, the offspring rats of each group (ten rats per group) underwent intravenous glucose tolerance tests (IVGTT) and the measurement of in vivo insulin action with the glucose–insulin clamp technique. After the clamp tests, the rats were killed by CO2 asphyxiation and their pancreases were quickly collected. Pancreases (six rats per group) were fixed with 10 % formalin in PBS for histology; other pancreases (four rats per group) were cut into small pieces (about 1 mm3) and fixed in 2·5 % glutaraldehyde for electron microscopic analysis. The offspring rats from each group (ten rats per group) that did not undergo the glucose tolerance test and clamp test were killed by CO2 asphyxiation after an overnight fast, and their pancreases prepared for islet isolation. The studies were carried out under the guidelines for the use and care of laboratory animals and were approved by the Institutional Animal Care and Use Committee of the Korea Institute of Radiological and Medical Sciences.
In vivo glucose-induced insulin secretion tests and euglycaemic–hyperinsulinaemic clamp studies
IVGTT and euglycaemic–hyperinsulinaemic clamp studies were performed on awake and unstressed rats, aged 20 weeks, 12 h after food withdrawal as previously described(Reference Park, Jin and Cho16). Each rat underwent catheter placement into two tail veins and a tail artery for infusion and blood sampling, respectively. For IVGTT, a single injection of glucose (0·5 g glucose/kg body weight) was administered via a tail vein. Blood samples were collected sequentially from the tail artery before (0 min) and 2, 6, 10, 20 and 50 min after glucose administration. Plasma glucose concentration was immediately determined by the glucose oxidase method (YSI 2300; YSI Inc., Yellow Springs, OH, USA) and plasma insulin concentration was measured by a RIA test using a rat insulin kit (Linco Research, St Charles, MO, USA). For euglycaemic-hyperinsulinaemic clamp studies, a continuous intravenous infusion of purified human insulin (Novolin R; Novo Nordisk, Bagsvaerd, Denmark) was started at the rate of 72 pmol/kg per min and continued for 120 min with a syringe pump (Razel Scientific Instruments Inc., St Albans, VA, USA) through a tail vein. Blood samples were taken from the tail artery and glucose concentration was immediately determined at 10 min intervals. Blood samples of 200 μl were collected 60 and 120 min after insulin infusion for the determination of plasma insulin concentrations. A 25 % dextrose solution was infused through the other venous line at variable glucose infusion rates to maintain plasma glucose at basal levels.
Histological examination and islet morphometry
Formalin-fixed paraffin-embedded pancreases were cut into 4 μm thick sections and stained with haematoxylyin–eosin. The numbers of islets and the area of each islet were measured in every 20th section; five slides from each pancreas were analysed. The islet area was measured by planimetry using Leica Q Win image analysis software (Leica, Cambridge, Cambs, UK) at a magnification of 40 × .
Electron microscopy of mitochondria
Small pieces (approximately 1 mm3) of pancreas from the rats were fixed in 2·5 % glutaraldehyde and then post-fixed in 1 % osmium tetroxide and embedded in araldite (epoxy resin). Suitable areas for ultra-structural study were chosen after examining semi-thin sections under a light microscope. Ultra-thin sections were stained with uranyl acetate and lead citrate. The grids were examined under a Hitachi 600 electron microscope (Hitachi, Tokyo, Japan).
Immunohistochemical staining and its assessment
Immunohistochemical staining for respiratory chain complex II 70 kDa subunit and complex IV (or cytochrome c oxidase; COX) subunit I of pancreas was performed. The Zymed non-biotin amplification system (Zymed, Carlsbad, CA, USA) was used. The primary antibodies used were mouse monoclonal antibodies against respiratory chain complex II 70 kDa subunit (1:1000 dilution; Molecular Probes, Eugene, OR, USA), complex IV (COX) subunit I (COX I) (1:1000 dilution; Molecular Probes), mitochondrial transcription factor A (Tfam) (1:1000 dilution; GenWay Biotech Inc., San Diego, CA, USA), insulin (1:200 dilution; Dako, Carpinteria, CA, USA) and glucagon (1:1000 dilution; Dako). For these antibodies, the sections were pretreated with 0·01 m-sodium citrate butter (pH 6·0) and autoclaved for 1 min (at 121°C) to retrieve the antigen. Immunoreactivity of the pancreatic islet was recorded by a semi-quantitative grading system considering both the intensity of staining and the proportion of stained cells. The intensity was recorded as 0 (no staining), 1+(weak), 2+ (moderate) and 3+ (strong staining). The fraction of positive cells in the islets was recorded (0–100 %). The total score of staining was calculated as multiplication of the staining intensity score and fraction of positive cells. Scores above 150 were defined high expression; below 150 were considered low. The staining was evaluated by two independent expert pathologists in a blind manner.
Islet isolation and Western blot
Islets were isolated by collagenase digestion. Briefly, the pancreas was inflated with solution containing collagenase P (Roche, Basel, Switzerland), excised and maintained at 37°C for 20 min, and then vigorously shaken at 37°C for 14 min. After a quick spin, the tissue pellet was washed twice with 10 ml cold Hank's balanced salt solution (HBSS), passed through a 925 μm Spectra mesh filter (Fisher, St Louis, MO, USA) to remove large debris, and re-suspended in 5 ml of 25 % Ficoll® (type 400-DL; Sigma-Aldrich Corp.) prepared with HBSS in a 50 ml conical tube. Then, 2·5 ml of 23, 20·5 and 11 % Ficoll® were layered carefully on top of each other, and the gradient was centrifuged for 15 min at 800 g. Layers above the 25 % Ficoll® containing the isolated islets were collected and washed with HBSS, and the islets were pelleted by a 5 min centrifugation at 800 g. The islets were handpicked under stereomicroscopic observation. The expression of respiratory chain complex II 70 kDa subunit and COX I was examined by Western blot analyses. Briefly, islet cells were lysed in a buffer supplemented with a protease inhibitor cocktail (Sigma-Aldrich Corp.) and 0·1 mm-phenylmethanesulfonyl fluoride (Sigma-Aldrich Corp.). After insoluble materials were removed by centrifugation, samples containing 20 μg protein were mixed with loading buffer for separation using SDS-PAGE. After transfer to nitrocellulose membranes which were blocked with 5 % non-fat dry milk in TTBS (10 mm-2-amino-2-hydroxymethyl-propane-1,3-diol (Tris), 150 mm-NaCl, 0·5 % Tween 20) for 1 h, they were incubated with the antibodies for COX I and complex II (diluted 1:1000 in TTBS plus 3 % non-fat dry milk; mouse monoclonal antibodies; Molecular Probes) for 2 h. After washing the membranes, they were incubated with horseradish peroxidase-conjugated anti-mouse antibody (diluted 1:2000 in TTBS plus 3 % non-fat dry milk; Cell Signaling Technology, Inc., Danvers, MA, USA) for 1 h at room temperature. Antibody binding sites were visualised using an enhanced chemiluminescence blotting detection system (Amersham, Little Chalfont, Bucks, UK).
Analysis and statistics
Experimental results are given as mean values with their standard errors. Statistical analyses were performed using non-parametric Kruskal–Wallis one-way ANOVA for comparisons of unpaired data between the three groups. Individual between-group comparisons were performed using the Mann–Whitney U test after ANOVA. Differences were considered significant at P < 0·05.
Results
Body weights
As previously reported(Reference Park, Jin and Cho16, Reference Park, Kim and Kim17), body weights were lower in the LP group than in the control group for all ages examined. Having a normal diet after weaning was not sufficient for the catch up of normal body weight. Body weight was not different between the LP-T and LP groups (Table 1).
Table 1 Body weights of offspring (g)
(Mean values with their standard errors for twenty animals per group)
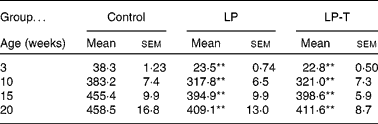
LP, low-protein diet; LP-T, taurine-supplemented low-protein diet.
** Mean value was significantly different from that of the control group (P < 0·01).
Insulin secretion and insulin sensitivity
IVGTT and euglycaemic–hyperinsulinaemic clamp tests were done at 20 weeks of age (Fig. 1). Fasting plasma glucose concentrations and glucose concentrations after intravenous glucose administration were not significantly different among the three groups. Fasting plasma insulin concentrations in the LP group tended to be lower than in the control group, but this was not statistically significant. The plasma insulin concentrations at 2, 6 and 50 min after glucose administration were significantly lower in the LP group than in the control group. Taurine supplementation tended to restore insulin response to glucose challenge, with plasma insulin concentrations of the LP-T group significantly higher than those of the LP group (P < 0·05) at 6 min (Fig. 1). The mean area under the curve (AUC) of insulin during IVGTT, which represents the insulin-secretory response, was reduced by 54·3 % in the LP group compared with the control group (P < 0·05). In this group, the early response (0–10min) was particularly blunted (P < 0·05; Table 2). The mean AUC (0–10 min) of the LP-T group was 40·7 % greater than in the LP group (P < 0·05), showing that this restoration was in the early insulin response. Whole-body insulin sensitivity was measured during hyperinsulinaemic clamp studies. The insulin sensitivity index was calculated from glucose infusion rates divided by steady-state plasma insulin concentrations. No significant difference in the insulin sensitivity index was noted among the three groups (Table 2).

Fig. 1 Changes in plasma glucose (a) and insulin (b) concentrations to intravenous glucose tolerance tests at 20 weeks of age in male offspring of rats fed a control diet (–□–; n 10), a low-protein (LP) diet (–○–, n 7) or a low-protein diet supplemented with taurine (LP-T; –▲–; n 10) during pregnancy and lactation. Values are means, with standard errors represented by vertical bars. * Mean value was significantly lower than that of the control group (P < 0·05). † Mean value was significantly higher than that of the LP group (P < 0·05).
Table 2 Results of intravenous glucose tolerance tests and euglycaemic–hyperinsulinaemic clamp studies undertaken at 20 weeks of age
(Mean values with their standard errors)
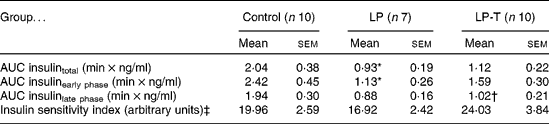
LP, low-protein diet; LP-T, taurine-supplemented low-protein diet; AUC, area under the curve.
* Mean value was significantly different from that of the control group (P < 0·05).
† Mean value was significantly different from that of the LP group (P < 0·05).
‡ Insulin sensitivity index was calculated from glucose infusion rate divided by steady-state plasma insulin concentration.
Histological examinations of the tissues and islet morphometry
The pancreases from six 20-week-old rats of each group were histologically examined. The exocrine and endocrine structures of the pancreas were well maintained in all three groups. In the LP group, the size of the islets was smaller and the number of islets was decreased compared with the control group. Taurine supplementation restored these changes. Morphometric analyses showed a decreased mean area and number of islets in the LP group, with restoration in the LP-T group (Table 3). When we performed immunostaining of the islets for insulin and glucagon, the fraction of insulin-positive cells was reduced and the fraction of glucagon-positive cells was increased in the LP group. These changes were restored in the LP-T group (Table 3).
Table 3 Numbers of islets per section, mean area of islets, and the fractions of immunoreactive cells for insulin and glucagon in the islets‡
(Mean values with their standard errors)
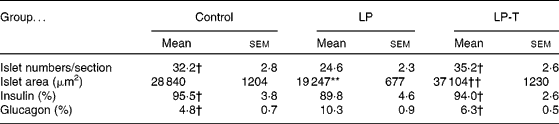
LP, low-protein diet; LP-T, taurine-supplemented low-protein diet.
** Mean value was significantly different from that of the control group (P < 0·01).
Mean value was significantly different from that of the LP group: † P < 0·05, †† P < 0·01.
‡ The pancreases from six rats per group were examined. The numbers of islets and the area of each islet were measured in every 20th section of 4 μm thick sections. Five slides from each pancreas were analysed. The numbers of total islets counted were 968 in the control group, 739 in the LP group and 1058 in the LP-T group, respectively.
Electron microscopic findings of mitochondria
The ultra-structural changes of mitochondria in islet cells of the pancreas were investigated at 20 weeks of age (Fig. 2). β-Cells of pancreatic islets showed changes in the LP group. Usually, β-cells of the pancreatic islet have highly characteristic secretory granules containing a crystalline electron-dense core, which are eccentric and surrounded by a large halo. In the control group, the electron microscopic analysis of β-cells showed no pathological changes. The cytoplasm of the normal β-cells was filled with numerous evenly dispersed secretory granules with electron-dense cores, and round- to oval-shaped mitochondria with relatively parallel linear cristae. The cores of the secretory granules were eccentric and relatively homogeneously electron dense. The halo between the core and external single membrane was large. In the LP group, the β-cells had a decreased number of secretory granules (513·2 (sem 46·9) in the control group, 314·2 (sem 27·6) in the LP group and 535·8 (sem 55·1) in the LP-T group, P < 0·01, analysed with five micrographs from four samples of each group; magnification 5000 × ). They also had abnormally shaped and sized secretory granules, with less dense cores. The less dense-cored secretory granules were immature β granules, which were slightly larger and had a narrower halo compared with mature secretory granules(Reference Bonner-Weir, Kahn, Weir and King25). Mitochondria were slender and elongated in appearance with indistinct cristae. The matrix was denser than that of normal ones. Some β-cells had increased number of mitochondria. In the LP-T group, the abnormalities of the mitochondria and secretory granules were restored and much closer to those of the normal β-cells of the control group.

Fig. 2 Electron micrographs of β-cells in islets of the control (C), low-protein diet (LP) and taurine-supplemented low-protein diet (LP-T) groups. While β-cells of the control group contained numerous β granules with intact-appearing mitochondria, the LP group showed abnormal mitochondria in shape and size, which were slender and elongated in appearance with a decreased number of β granules. Half of the β granules also had a primitive appearance; with an enlarged size with pale cores and much less of a halo compared with the mature β granules. β-Cells from the LP-T group revealed a recovery of mitochondrial abnormalities and a near-normal number of β granules.
Immunohistochemical analysis and Western blot analysis of cytochrome c oxidase subunit I, complex II and mitochondrial transcription factor A
The mitochondrial electron transport chain consists of the partially mitochondrial-encoded complexes I, III and IV and the exclusively nuclear-encoded complex II. Also, mtDNA transcription and replication are regulated by Tfam(Reference Moraes26). We investigated the expression of COX I, which is a mitochondrial DNA-encoded protein, the 70 kDa subunit of complex II, which is a nuclear-encoded mitochondrial protein, and Tfam, which is a nuclear-encoded protein in the pancreas at 20 weeks of age. The results of the immunohistochemical staining are shown in Fig. 3 and Table 4. The immunoreactivity of COX I in the islets was weaker in the LP group compared with the control group (P < 0·05). In the islets of the LP-T group, the reactivity of COX I was almost the same as that of the control group, meaning that COX I expression was restored by taurine supplementation. The immunoreactivity of complex II was much stronger in the islet than the acinar cells, with immunoreactivity in the islet not significantly different among the three groups. The immunoreactivity of Tfam in the islets also was not significantly different among the three groups (Fig. 3; Table 4).

Fig. 3 Immunohistochemical staining for cytochrome c oxidase subunit I (COX I), complex II and mitochondrial transcription factor A (Tfam) in the pancreas of the control (C), low-protein diet (LP) and taurine-supplemented low-protein diet (LP-T) groups. The immunoreactivity of COX in the pancreatic islets was lighter in the LP group than in the control group, and this was restored in the LP-T group ( → ). However, the immunoreactivities of complex II and Tfam were not different among the three groups. Magnification is × 200 for all images.
Table 4 Semi-quantitative demonstration of immunochemical staining for cytochrome c oxidase subunit I (COX I), complex II and mitochondrial transcription factor A (Tfam) in the islets*

LP, low-protein diet; LP-T, taurine-supplemented low-protein diet.
* The total score of staining was calculated as multiplication of the staining intensity score and the fraction of positive cells. Scores above 150 were defined as high expression; scores below 150 were considered low.
Western blot analysis of the isolated islet cells showed that the expression of COX I was decreased in the LP group and restored in the LP-T group. The expression of complex II was not different among the three groups (Fig. 4).

Fig. 4 Western blot (a) and densitometric analyses (b) of isolated islet cells. The expression of cytochrome c oxidase subunit I (COX I) was decreased in the low-protein diet (LP; ![]() ) group compared with the control (C; □) group and was restored in the taurine-supplemented low-protein diet (LP-T; ■) group. The expression of complex II was not different among the three groups.
) group compared with the control (C; □) group and was restored in the taurine-supplemented low-protein diet (LP-T; ■) group. The expression of complex II was not different among the three groups.
Discussion
In the present study, we demonstrated that protein malnutrition early in life induces abnormal mitochondrial changes that are present later in life, with all these changes restored by taurine supplementation. The shape and size of the mitochondria were abnormal in β-cells, and the expression of the mtDNA-encoded COX I gene in islet cells was decreased. This was associated with a decreased number of insulin-secretory granules in β-cells, a decreased fraction of β-cells in the islets, reduced number and size of pancreatic islets, as well as a decreased insulin-secretory response in vivo.
Despite the substantial epidemiological studies that revealed links between intra-uterine growth retardation and susceptibility to diabetes in adult life, the mechanisms underlying the pathophysiology of these associations are not yet known. In regards to the pathogenesis of the thrifty phenotype, mitochondrial dysfunction has been proposed as a link between poor nutrition early in life and diabetes as an adult(Reference Lee15). Intra-uterine growth retardation due to the ligation of the uterine arteries induced mitochondrial dysfunction and increased reactive oxygen species production in the fetal β-cell(Reference Simmons, Suponisky-Kroyter and Selak27). Our previous studies showed that protein malnutrition during gestation and lactation decreased mtDNA content and the mtDNA-encoded gene expression of the liver and skeletal muscle(Reference Park, Kim and Kim17). These rats also had decreased mtDNA content in the pancreas accompanied by decreased pancreatic β-cell mass, and reduced insulin-secretory responses to a glucose load(Reference Park, Jin and Cho16). The results of the present study offer more evidence that strongly support the suggestion that mitochondrial dysfunction is a pathogenic link between poor nutrition early in life and diabetes as an adult.
The present study demonstrated for the first time that taurine supplementation to the protein-malnourished rats restores the morphological deterioration of mitochondria and the expression of COX I of the offspring. The mechanisms of how taurine affects the mitochondria are not yet known. The cellular actions of taurine are numerous, including the regulation of cell volume, extracellular and intracellular Ca mobilisation, its role as an antioxidant, and inhibition of apoptosis(Reference Huxtable19). Immunocytochemical studies have demonstrated that taurine immunoreactivity increases in the mitochondria, indicating taurine localisation(Reference Lobo, Alonso and Martin del Rio28, Reference Terauchi and Nagata29). Suzuki et al. (Reference Suzuki, Suzuki and Wada30) found two novel taurine-containing modified uridines in mtDNA, and showed that taurine was a constituent of mitochondrial tRNA. A lack of these modifications, which has been found in the patients of mitochondrial encephalomyopathies, causes defective translation which might significantly contribute to the defective mitochondrial function in mitochondrial diseases. According to these findings, we can speculate that low taurine in the fetus by intra-uterine malnutrition may induce a deficiency of modification of nucleosides that leads to defective translation activity and mitochondrial dysfunction in the thrifty phenotype. Another possible mechanism is increased oxidative stress because of taurine deficiency. Protein malnutrition is associated with depressed antioxidant defence systems and increased oxidative stress(Reference Huang and Fwu31). Proteome analysis of fetal protein-malnourished pancreases revealed that antioxidant protein 2, which protects the pancreas against oxidative injury by reducing H2O2, was down-regulated(Reference Sparre, Reusens and Cherif32). Cells during development, especially β-cells, have a high energy requirement and poor antioxidant defence mechanism(Reference Tiedge, Lortz and Drinkgern33), and mitochondria are highly vulnerable to oxidative stress because they are the main site of free radical formation. Taurine is known to be an antioxidant. It has been suggested that it protects against oxidative damage of cellular membrane structures by removing the extremely reactive and oxidative compound hypochlorite, and by decreasing rates of malondialdehyde formation from unsaturated membrane lipids(Reference Huxtable19). From these suggested mechanisms, we can speculate that taurine restores the malnutrition-associated mitochondrial changes by protecting mitochondria from oxidative stress. Indeed, taurine has been found to protect against myocardial mitochondrial injury induced by hyperhomocysteinaemia in rats(Reference Chang, Xu and Yu34), and prevent tamoxifen-induced mitochondrial oxidative damage in mice(Reference Parvez, Tabassum and Banerjee35). Taurine can also act as a pH buffer in the mitochondrial matrix and stabilise mitochondrial oxidative phosphorylation(Reference Hansen, Andersen and Birkedal36), which can be another possible mitochondrial protective mechanism of taurine.
The present study demonstrated that taurine supplementation to protein-malnourished rats restores the morphological changes of mitochondria of the offspring. Mitochondrial biogenesis is a poorly understood process. In patients with mitochondrial encephalomyopathies caused by pathogenic mtDNA mutations, mitochondrial morphological changes usually present together with mitochondrial dysfunction(Reference Bonilla and Tanji37), and the mitochondrial morphology in the present study is very similar to that of patients with mitochondrial encephalomyopathies(Reference Bonilla and Tanji37). It is very hard to conclude that mitochondrial morphological change is a cause or consequence, or just an epi-phenomenon of mitochondrial dysfunction in the present study. But when we consider that the genes involved in mitochondrial biogenesis are all nuclear encoded, we can surmise very intimate coordination between the nuclear and mitochondrial genome in the programming of the thrifty phenotype.
In the present study, the offspring of protein-malnourished rats showed decreased pancreatic islet mass, and this was restored by taurine supplementation. Poor intra-uterine nutrition is known to be associated with impaired β-cell development(Reference Snoeck, Remacle and Reusens6, Reference Garofano, Czernichow and Breant38). Decreased β-cell mass seems to be associated with decreased rates of β-cell proliferation, impaired differentiation or increased rates of apoptosis(Reference Petrik, Reusens and Arany8, Reference Garofano, Czernichow and Breant38). Taurine is necessary for normal development. It has trophic effects in the developing endocrine pancreas. In previous studies, taurine supplementation of a maternal low-protein diet showed complete restoration of islet cell proliferation and apoptosis(Reference Boujendar, Reusens and Merezak22), islet vascularisations(Reference Boujendar, Arany and Hill23) and the insulin secretion of the offspring in vitro (Reference Cherif, Reusens and Ahn24).
The present study showed that the reduced insulin-secretory response of the LP group was restored by taurine supplementation especially in early-phase secretion. For the secretion of insulin from β-cells, an increase of intracellular ATP through mitochondrial oxidative phosphorylation of glucose is necessary(Reference Gerbitz, van den Ouweland and Massen39). This is followed by the closure of the ATP-sensitive K+ channel and increased intracellular Ca2+, which eventually triggers insulin secretion. The early phase of insulin secretion is absolutely dependent on the intracellular Ca2+ concentration, but the late phase of secretion seems not to depend solely on the intracellular Ca concentration(Reference Henquin40). Therefore, the defect in the early phase of secretion in the LP group in the present study was due to the lack of a rapid increase in intracellular Ca2+; this can be explained by insufficient ATP generation due to mitochondrial dysfunction. The loss of early-phase secretion to glucose has been considered to be an early sign of β-cell dysfunction, characteristic of type 2 diabetes(Reference Cerasi, Luft and Efendic41). In the LP group of the present study, secretory granules of β-cells were decreased in number and showed morphological characteristics of immature granules. The maturation process of proinsulin-containing granules involves the acidification of the granules, which is due to activation of an ATP-dependent proton pump. This is critical for the conversion of proinsulin to insulin(Reference Rhodes, Shoelson, Halban, Kahn, Weir and King42). Lack of ATP production by mitochondrial dysfunction in β-cells could be a main cause of low insulin secretion in the fetal malnourished rat.
A recent report provides strong evidence that supports the suggestion that mitochondrial dysfunction is a pathogenic link in the thrifty phenotype(Reference Reusens, Sparre and Kalbe43). Microarray analysis of the pancreas revealed that one of the most affected pathways by fetal protein malnutrition was cellular respiration: genes encoded for the tricarboxylic acid cycle and mitochondrial proteins were affected. Furthermore, those islets were unable to enhance their ATP production when stimulated with glucose. Maternal taurine supplementation normalised the expression of all altered genes and ATP production(Reference Reusens, Sparre and Kalbe43).
In the present study, we showed the abnormal shape and number of mitochondria, and the reduced expression of mtDNA-encoded COX I due to fetal protein malnutrition. However, the expression of Tfam was not different among the three groups. This may be part of a compensatory process in response to a decreased expression of COX I. Additional analyses to measure direct mitochondrial function, such as ATP production, would strongly substantiate mitochondrial dysfunction.
In conclusion, we have presented evidence of the pathogenic role of mitochondria in the development of the thrifty phenotype and the role of taurine in mediating these mitochondrial changes. Although fetal or neonatal malnutrition in animals may not always adequately represent the human situation, taurine supplementation could be a possible method to prevent deleterious alterations (programming) in early life due to nutritional deprivation. Further studies will be required to elucidate the mechanism by which fetal protein malnutrition causes the long-lasting mitochondrial changes, and how taurine mediates these changes.
Acknowledgements
The present study was partially supported by a grant from the Korea Institute of Radiological and Medical Sciences (no. KIRAMS/RR-023-2005).
Y. Y. L., K. S. P. and H. K. L. designed the research; H.-J. L., S.-S. L., J. S. K., C. J. J. and S.-H. P. conducted the research; K. H. Y. helped to analyse the data and write the paper; Y. Y. L. analysed the data, wrote the paper, and had primary responsibility for the final content.
The authors declare that there are no potential conflicts of interest relevant to this paper.








