


Several lines of evidence indicate that a disproportionate burden of preventable disease, death and disability exists in racial and ethnic minority populations, especially African Americans in the USA. Differences in the prevalence of the metabolic syndrome have been noted in the US National Health and Nutrition Examination Survey (NHANES) studies, with prevalence notably higher among African American women(Reference Barnes, Grant and Hansel1). In addition, the profile of the metabolic syndrome differs among ethnicities, with African Americans showing a smaller contribution of dyslipidaemia (i.e. fewer HDL-cholesterol and TAG abnormalities) compared with European Americans(Reference Olshansky, Passaro and Hershow2). However, there is greater insulin resistance among African Americans, even during childhood(Reference Vaidya, Szklo and Ding3). Likewise, CVD risk shows significant racial and ethnic differences, with the highest age-adjusted death rates observed in African Americans(Reference Burns, Lee and Arslanian4). Differences in the prevalence and severity of chronic diseases involving inflammation are further corroborated by differences in inflammatory biomarkers, including C-reactive protein(Reference Bjorksten, Clayton and Ellwood5, Reference Khera, McGuire and Murphy6). The striking racial and ethnic differences in the prevalence and/or severity of common diseases is probably explained by a complex combination of environmental, social, cultural or economic factors, and genetic factors are likely to be very important as well(Reference Albert, Glynn and Buring7).
Agricultural and industrial revolutions have increased the quantity and variety of foods but have not necessarily improved the human diet(Reference Cordain, Eaton and Sebastian8, Reference Flinn, Geary and Ward9). In fact, more than 70 % of the energy consumed by humans today in developed countries would not have been available to our hunter–gather ancestors(Reference Cordain, Eaton and Sebastian8). This rapid shift in the type of energy consumed by modern humans appears to have created a sort of malnutrition in developed countries whereby certain nutrients are not well tolerated by our ‘hunter–gather’ genes. This problem has become a global issue as Western-derived food supplies and practices expand with global trade. The negative impact of the modern diet on health is likewise exported to developing nations.
Humans can synthesise a wide range of fatty acids, but they lack key enzymes (Δ12 and Δ15 desaturases) necessary to synthesise the initial PUFA used in the key PUFA biosynthetic pathway in mammals(Reference Innis10, Reference Innis11). Therefore, linoleic acid (18 : 2n-6; LA) and α-linolenic acid (18 : 3n-3) are essential fatty acids(Reference Innis10). Once obtained from the diet, they are converted to long-chain PUFA (LC-PUFA) by the alternate actions of two fatty acid desaturase (FADS) enzymes (Δ6 and Δ5 desaturases encoded for by FADS2 and FADS1, respectively) and an elongase enzyme that introduce carbon–carbon double bonds and increases chain length by two carbons, respectively(Reference Guillou, Zadravec and Martin12). Additionally, preformed LC-PUFA such as arachidonic acid (20 : 4n-6; AA) can also be readily obtained from human diets. AA is found in relatively high concentrations in the meats of grain-fed animals and eggs(Reference Cordain, Eaton and Sebastian8, Reference Blasbalg, Hibbeln and Ramsden13).
Once produced, AA and its metabolic products play important roles in orchestrating immunity and inflammation(Reference Schmitz and Ecker14–Reference Torrejon, Jung and Deckelbaum16) via their ability to directly affect normal and pathophysiological responses through: (i) conversion to potent AA-derived bioactive products (including PG, thromboxanes, leukotrienes and lipoxins); and (ii) regulation of cellular receptors (NFκB(Reference Novak, Babcock and Jho17, Reference Soberman and Christmas18), PPAR(Reference Jump and Clarke19) and sterol regulatory element-binding protein-1c(Reference Xu, Nakamura and Cho20, Reference Worgall, Sturley and Seo21)), thereby modulating the expression of many genes that control immune responses (cytokines such as IL-1, IL-6, IL-12 and TNF-α; chemokines such as IL-8, macrophage inflammatory protein-1a and monocyte chemotactic protein-1; adhesion molecules such as intercellular adhesion protein and E-selectin; and inducer effector enzymes such as inducible NO synthase and cyclo-oxygenase-2(Reference Ghosh and Karin22)).
During the 20th century in the USA, there has been a dramatic increase in LA consumption (from an estimated 2·8 % to nearly 8 % of energy)(Reference Blasbalg, Hibbeln and Ramsden13) primarily as a result of increased availability of soyabean oil, margarine and poultry(Reference Blasbalg, Hibbeln and Ramsden13). In fact, nearly 85 % of total PUFA in a typical Western diet is LA(Reference Muskiet, Montmayeur and le Coutre23). Biochemical studies using stable isotopes largely carried out in subjects of European ancestry indicate that only a small proportion of dietary LA (about 0·2 %) can be converted to AA in humans(Reference Hussein, Ah-Sing and Wilkinson24). However, humans do appear to be able to synthesise sufficient AA or extract AA from the diet to maintain AA status(Reference Vlaardingerbroek, Hornstra and de Koning25). Nevertheless, this low rate of conversion has been assumed to apply to all human populations equally. For example, the Advisory Committee from the American Heart Association has concluded ‘that at least 5 to 10 % of energy from [n]-6 PUFAs reduces the risk of CHD relative to lower intakes’(Reference Harris, Mozaffarian and Rimm26). This conclusion was based on several assumptions, one of which concluded that ‘wide variations in dietary LA (above minimal essential intakes) do not materially alter tissue AA content’(Reference Harris, Mozaffarian and Rimm26). Yet the literature suggests that high LA-containing diets can reduce the LC-PUFA content of tissues when modelled from per capita food consumption data(Reference Blasbalg, Hibbeln and Ramsden13) or by direct analysis of tissues(Reference Friesen and Innis27).
Studies over the past 5 years suggest that there is probably large genetic variability in the rate of conversion of LA to AA(Reference Malerba, Schaeffer and Xumerle28–Reference Rzehak, Heinrich and Klopp32). Importantly, genetic variants that are associated with higher levels of AA are also associated with elevated levels of markers of systemic inflammation and the incidence of certain inflammatory disorders(Reference Martinelli, Girelli and Malerba31). The present study examines levels of LA and AA, their association with genetic variants in the FADS gene cluster, and the frequency of high-converting genotypes in patients of African and European ancestry with diabetes or the metabolic syndrome. The results of the present study demonstrate that there is a marked increase in AA and in the frequency of alleles that favour AA synthesis in African Americans with diabetes or the metabolic syndrome. The present study reveals that there are probably differential effects of high concentrations of LA in African and European American populations in the USA and caution should be exercised with regard to dietary recommendations that assume n-6 PUFA metabolism is uniform in all human populations.
Experimental procedures
Subjects
The study population was derived from the Diabetes Heart Study (DHS; n 229) and included European American (n 166; from eighty-nine families) and African American (sixty-three from thirty-three families) subjects with diabetes or the metabolic syndrome. Methods for ascertainment and recruitment for the DHS have been described previously(Reference Bowden, Lehtinen and Ziegler33). Briefly, siblings concordant for type 2 diabetes mellitus without renal insufficiency were recruited, along with unaffected siblings. The metabolic syndrome was defined using the standard definition from the Executive Summary of The Third Report of The National Cholesterol Education Program Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III)(34). The Wake Forest University School of Medicine Institutional Review Board approved study protocols, and all participants provided written informed consent.
Fatty acid analysis
Serum was isolated from fasting whole blood samples. Fatty acid methyl esters were prepared(Reference Metcalfe, Schmitz and Pelka35) in duplicate from serum samples (100 μl) in the presence of an internal standard (triheptadecanoin; NuChek Prep, Elysian, MN, USA) as previously described in detail(Reference Weaver, Ivester and Seeds36). A panel of twenty-three fatty acids was quantified by GC with flame ionisation detection. Individual fatty acids are expressed as percentage of total fatty acids in a sample. For all samples, data peaks on chromatograms were examined to ensure peak quality and consistency of retention times. Fatty acids in samples were identified based on the retention times of methyl ester derivatives of authenticated fatty acid standards. These standards included Supelco 37 Component FAME Mix (Supleco, Bellefonte, PA, USA) and other individual methyl ester derivatives from Supelco (oleate, cis-11-vaccenoate, linoleate, eicosapentaenoate, n-3 docosapentaenoate), Cayman Chemicals (Ann Arbor, MI, USA; stearidonate), Matreya (Pleasant Gap, PA, USA; eicosadienoate, dihomo-γ-linolenoate, arachidonate) and NuChek Prep (cis-eicosatrienoate, docosadienoate, docosatetraenoate, n-6 docosapentaenoate, docosahexaenoate, tricosanoate, tetracosanoate). Product:precursor ratios of circulating fatty acids, an estimate of enzymic activity, were calculated from fatty acid mass data.
For statistical analyses of fatty acid data, normal kernel density estimation (implemented in S-Plus; TIBCO Software Inc., Palo Alto, CA, USA) was used to obtain estimates of the probability density functions. Linear mixed models were used to assess the racial difference in the fatty acids and ratios adjusting for sex and age. Family was treated as a random effect and age, sex and race as fixed effects. Residuals were examined to assess the model assumptions.
Genotyping and tests for association in the Diabetes Heart Study subjects
Seven SNP mapping to the FADS gene cluster (rs174537, rs102275, rs174546, rs174556, rs1535, rs174576, rs174579) were selected based on previous publications(Reference Schaeffer, Gohlke and Muller30, Reference Tanaka, Shen and Abecasis37, Reference Allayee, Baylin and Hartiala38). Genotypes were determined using a Sequenom MassARRAY SNP genotyping system (Sequenom Inc., San Diego, CA, USA)(Reference Buetow, Edmonson and MacDonald39). Of the samples, 3·5 % were genotyped in duplicate with 100 % reproducibility across the SNP. Linkage disequilibrium (LD) was assessed by calculating D' and r 2 within Haploview(Reference Barrett, Fry and Maller40) relying on a set of independent individuals in the data (a random selection of a single individual from each pedigree, n 33 and n 89 African American and European American subjects, respectively) and haplotype blocks were defined according to the algorithm of Gabriel et al. (Reference Gabriel, Schaffner and Nguyen41).
Allele and genotype frequencies for each SNP were calculated from unrelated probands and tested for departure from Hardy–Weinberg equilibrium using a χ2 goodness-of-fit test. Associations between SNP and traits were performed using a series of variance components measured genotype models as implemented in Sequential Oligogenic Linkage Analysis Routines (SOLAR)(Reference Almasy and Blangero42). Significance was evaluated using the likelihood ratio tests based on the correlation structure suggested by the familial relationships. The additive genetic model was the primary model of interest; however, for SNP with less than ten individuals homozygous for the minor allele a dominant model was analysed and all models included age and sex covariates. When necessary, phenotypes included in these analyses were transformed using the natural logarithm.
Allele frequency analysis of global populations
Publicly available data for ten populations from the International HapMap project (phase III; http://www.hapmap.org) were used to derive allele frequencies for the rs174537 SNP, which was among those that were genotyped in the DHS population. These data include 1046 samples from the following ten populations: Luhya population in Kenya (LWK; n 110), Yoruba population in Nigeria (YRI; n 147), Mexican ancestry in Los Angeles, CA, USA (MEX; n 58), African American population in the Southwest USA (ASW; n 57), Gujarati Indian population in Houston, TX, USA (GIH; n 101), Japanese population in Tokyo (JPT; n 113), Han Chinese population in Beijing (CHB; n 137), Chinese population in Denver, CO, USA (CHD; n 108), Italian population in Tuscany (TSI; n 102) and a European American population in Utah State, USA (CEU; n 113).
Results
Serum fatty acid profiles differ in Americans of African and European descent
Fig. 1(a)–(d) show the distribution of n-6 PUFA in the sera of African American (n 63, 41·3 % male, aged 61·0 (sd 10·1) years) and European American (n 166, 42·7 % male, aged 68·2 (sd 10·5) years) adults with diabetes or the metabolic syndrome from the DHS(Reference Bowden, Lehtinen and Ziegler33). There was a pronounced enhancement in levels of serum AA (P = 2·29 × 10− 9) in African American compared with those in European American subjects. In contrast, no differences were observed in the levels of LA, γ-linolenic acid (GLA) or dihomo-γ-linolenic acid (DGLA), all precursors of AA. These data suggest that similar levels of LA are ingested by both populations.

Fig. 1 (a)–(d) Serum fatty acid distributions of n-6 PUFA in African American (AfAm; n 63; 41·3 % male; age 61·0 (sd 10·1) years; ![]() ) and European American (EAm; n 166; 42·7 % male; age 68·2 (sd 10·5) years;
) and European American (EAm; n 166; 42·7 % male; age 68·2 (sd 10·5) years; ![]() ) adults with diabetes or the metabolic syndrome from the Diabetes Heart Study. Normal kernel density estimation (implemented in S-Plus; TIBCO Software Inc., Palo Alto, CA, USA) was used to obtain estimates of the probability density functions that show the distribution of subjects having circulating n-6 PUFA as a function of percentage of total fatty acids by race. (a), Linoleic acid (LA), P = 0·861; (b), γ-linolenic acid (GLA), P = 0·222; (c), dihomo-γ-linolenic acid (DGLA), P = 0·534; (d), arachidonic acid (AA), P = 2·29 × 10− 9. (e)–(g) The product:precursor ratios of circulating fatty acids were used to estimate (e) fatty acid desaturase 2 (FADS2; GLA:LA ratio; P = 0·348), (f) elongase (DGLA:GLA ratio; P = 0·422) and (g) fatty acid desaturase 1 (FADS1; AA:DGLA ratio; P = 1·44 × 10− 5) enzymic efficiencies for the AfAm (
) adults with diabetes or the metabolic syndrome from the Diabetes Heart Study. Normal kernel density estimation (implemented in S-Plus; TIBCO Software Inc., Palo Alto, CA, USA) was used to obtain estimates of the probability density functions that show the distribution of subjects having circulating n-6 PUFA as a function of percentage of total fatty acids by race. (a), Linoleic acid (LA), P = 0·861; (b), γ-linolenic acid (GLA), P = 0·222; (c), dihomo-γ-linolenic acid (DGLA), P = 0·534; (d), arachidonic acid (AA), P = 2·29 × 10− 9. (e)–(g) The product:precursor ratios of circulating fatty acids were used to estimate (e) fatty acid desaturase 2 (FADS2; GLA:LA ratio; P = 0·348), (f) elongase (DGLA:GLA ratio; P = 0·422) and (g) fatty acid desaturase 1 (FADS1; AA:DGLA ratio; P = 1·44 × 10− 5) enzymic efficiencies for the AfAm (![]() ) and the EAm (
) and the EAm (![]() ) populations. Values are means, with standard deviations represented by vertical bars. Linear mixed models were used for statistical analyses to assess the racial difference in the fatty acids and ratios adjusting for sex, age and familial relationships (see Methods).
) populations. Values are means, with standard deviations represented by vertical bars. Linear mixed models were used for statistical analyses to assess the racial difference in the fatty acids and ratios adjusting for sex, age and familial relationships (see Methods).
Fig. 1(e)–(g) show the ratios of products to precursors (GLA:LA, DGLA:GLA and AA:DGLA) of circulating fatty acids, which are surrogates of actual enzymic activity at each biochemical step. As suggested by fatty acid levels (Fig. 1(a)–(c)), the conversion of LA to GLA (FADS2 activity) and GLA to DGLA (elongase activity) are not different in subjects of African and European descent. However, there appears to be dramatically different conversion rates for DGLA to AA in the two populations through the Δ5 desaturase (FADS1 activity) step (Fig. 1(g)). African American subjects exhibited a markedly higher AA:DGLA ratio (P = 1·44 × 10− 5) compared with that in European Americans, suggesting a strikingly increased ability of the former to convert medium-chain (less than twenty carbons) PUFA to LC-PUFA.
FADS SNP associations and frequencies
Tests for association were performed with seven SNP mapping to the FADS gene cluster in DHS subjects. The pattern of association in the European American subjects was highly consistent with previous reports(Reference Tanaka, Shen and Abecasis37, Reference Lattka, Illig and Heinrich43). Specifically, the pattern in European Americans was in high LD for this region, with a single LD block (53 kb) that included the seven SNP across FADS1 and part of FADS2 (not shown). In contrast, no LD blocks were observed in the African Americans (not shown). The strength of association for AA ranged from 1·1 × 10− 6 to 8·9 × 10− 8 in the European American subjects (Table 1). Evidence for association was also observed (Table 1) for DGLA (7·5 × 10− 6 to 7·1 × 10− 7) and GLA (4·9 × 10− 6 to 9·8 × 10− 11).
Table 1 Tests of association for seven SNP in the fatty acid desaturase (FADS) loci and γ-linolenic acid (GLA), dihomo-γ-linolenic acid (DGLA) and arachidonic acid (AA) in African American (n 63) and European American (n 166) subjects from the Diabetes Heart Study

* P < 0·01, ** P < 0·001, *** P < 0·00 001.
No associations were observed in the African Americans subjects, which was probably due to the limited sample size of this sample coupled with the much lower allele frequency of the minor alleles. It is important to note that no African American individuals were found in this relatively small population that were homozygous for the minor allele of rs174537.
Genotypic effect of rs174537 on n-6 PUFA and fatty acid desaturase 1 enzymic efficiency
Fig. 2 shows the distributions of DGLA, AA and the AA:DGLA ratio by genotype at rs174537. The major allele (G) is associated with an increase in the mean serum level of AA and is consistent with an additive model in European Americans (Fig. 2(b)). In contrast to the case for AA, the allele (G) was associated with a decrease in mean levels of DGLA, the immediate precursor of AA (Fig. 2(a)), and is also consistent with an additive model in European American subjects. For the ratio of AA:DGLA, an estimate of FADS1 activity (Fig. 2(c)), it was observed that the common allele (G) appeared to be associated with an increased trait mean, i.e. increased enzymic efficiency. As mentioned above, it was not possible to compare levels of PUFA in those with the homozygous minor allele with those with the homozygous major allele as there were no African Americans found in this population that exhibited homozygous minor alleles (Fig. 2(d)–(f)). The mean AA levels in the African American population (GG 9·9 (sd 1·8), GT 9·2 (sd 2·2)) were significantly greater (GG P = 0·003, GT P = 0·0073; two-tailed t test) than those in the European American group (GG 8·5 (sd 2·1), GT 7·6 (sd 1·8)).

Fig. 2 Fatty acid trait distribution differences between European American (n 159; (a)–(c)) and African American (n 63; (d)–(f)) adults with diabetes or the metabolic syndrome from the Diabetes Heart Study based on genotype at rs174537. Each sample is represented by a single symbol for European Americans (![]() ,
, ![]() ,
, ![]() ) or for African Americans (
) or for African Americans (![]() ,
, ![]() ) at each genotype. Sample means and 95 % CI for the sample mean are shown as the horizontal black line and bars, respectively. Fatty acid data are the percentage of total fatty acids and the arachidonic acid:dihomo-γ-linolenic acid ratio (AA:DLGA) was calculated from fatty acid mass. Genotypic data were unavailable for seven European American subjects. Mean value was significantly different from that for the GG genotype: *P < 0·01, ***P < 0·00 001 (two-tailed, pair-wise t test within populations). † Mean value was significantly different from that for the GT genotype (P < 0·05; two-tailed, pair-wise t test within populations).
) at each genotype. Sample means and 95 % CI for the sample mean are shown as the horizontal black line and bars, respectively. Fatty acid data are the percentage of total fatty acids and the arachidonic acid:dihomo-γ-linolenic acid ratio (AA:DLGA) was calculated from fatty acid mass. Genotypic data were unavailable for seven European American subjects. Mean value was significantly different from that for the GG genotype: *P < 0·01, ***P < 0·00 001 (two-tailed, pair-wise t test within populations). † Mean value was significantly different from that for the GT genotype (P < 0·05; two-tailed, pair-wise t test within populations).
FADS rs174537 frequencies differ in populations around the world
Striking differences were observed in the allele frequencies across a majority of these SNP in the FADS gene cluster between the African American and European American populations in the DHS (Table 2). The resultant genotypic frequencies for rs174537 were skewed toward the homozygous major allele (81 %: GG) in the African American population, with only 19 % heterozygous and no homozygous minor allele genotypes observed (Table 2). In contrast, the European American population exhibited a much lower frequency of the homozygous major allele (46 %), with 43 % heterozygous and 11 % with the homozygous minor allele. To evaluate the genotype distribution of these alleles on a global scale, patterns of genetic variation were examined within the FADS locus, and in particular in rs174537, in populations within The International HapMap Project. Fig. 3 shows striking differences in genotypic frequencies between different populations around the world, with the greatest differences observed between African populations from Kenya (LWK) and Nigeria (YRI) v. populations of Mexican ancestry living in Los Angeles, CA, USA (MEX). African Americans in the DHS have genotypic frequencies similar to those in another African American population in the Southwest USA (ASW) and a Gujarati Indian population in Houston, TX, USA (GIH). Greater than 75 % of individuals in each of these populations carry the major allele homozygous GG genotype. In contrast, our DHS European American population had frequencies similar to those of a Japanese population in Tokyo (JPT), a Chinese population in Beijing (CHB), an Italian population in Tuscany (TSI) and a European American population in Utah State, USA (CEU). Less than 50 % of individuals in these populations carry the major allele homozygous GG genotype.
Table 2 Genotypic distribution at rs174537 in Diabetes Heart Study populations
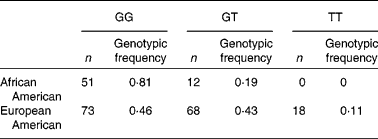

Fig. 3 Distributions of rs174537 genotype frequency in ten HapMap-derived global populations and the Diabetes Heart Study (DHS) African American (AfAm) and European American (EAm) subpopulations ( ↑ ), ranked from low to high for the homozygous major allele (GG, ![]() ). (
). (![]() ), TT; (
), TT; (![]() ), GT; MEX, Mexican ancestry from Los Angeles, CA, USA; CHD, Chinese population in Denver, CO, USA; CHB, Han Chinese population in Beijing; CEU, European American population in Utah State, USA; JPT, Japanese population in Tokyo; TSI, Italian population in Tuscany; GIH, Gujarati Indian population in Houston, TX, USA; ASW, African American population in the Southwest USA; LWK, Luhya population from Kenya; YRI, Yoruba population from Nigeria.
), GT; MEX, Mexican ancestry from Los Angeles, CA, USA; CHD, Chinese population in Denver, CO, USA; CHB, Han Chinese population in Beijing; CEU, European American population in Utah State, USA; JPT, Japanese population in Tokyo; TSI, Italian population in Tuscany; GIH, Gujarati Indian population in Houston, TX, USA; ASW, African American population in the Southwest USA; LWK, Luhya population from Kenya; YRI, Yoruba population from Nigeria.
Discussion
Multi-factorial diseases of chronic inflammation disproportionately affect African Americans in industrialised settings such as the USA(44), yet appear to be rare in continental Africans. Only 1–2 % of Africans on the African continent have type 2 diabetes, whereas the incidence is 11–13 % in individuals of African descent in industrialised nations consuming a Western diet(45, 46). It is clear that a complex interplay between genes and environment is contributing to these differences.
Several lines of evidence from the present study suggest that this interplay is likely to be important with regard to the dietary consumption and metabolism of n-6 PUFA. First, African Americans had higher levels of circulating AA compared with those of European descent. Importantly, there were no differences in levels of the levels of fatty acid precursors (LA and GLA) to n-6 LC-PUFA in these two populations of patients with diabetes or the metabolic syndrome. To date, few studies have examined the impact of ancestry on LC-PUFA synthesis and levels. In 1991, Horrobin et al. (Reference Horrobin, Ells and Morse-Fisher47) observed that AA levels in plasma phospholipids of nineteen subjects from Zimbabwe Africa were approximately 2-fold higher than those in a much larger group (n 458) of subjects with European ancestry. The present study strongly suggests that populations of African ancestry also have higher levels of circulating AA compared with those of European ancestry.
Second, there were marked differences between the African American and European American populations in the present study with regard to frequencies of alleles in several SNP in the FADS gene cluster, which have shown to be important in determining fatty acid levels. Specifically, rs174537 is the SNP near FADS1 that Tanaka et al. (Reference Tanaka, Shen and Abecasis37) have demonstrated to be most associated with AA levels (P = 5·95 × 10− 46). They demonstrated that individuals who were homozygous for the minor allele had significantly lower AA levels compared with those who carried the homozygous major allele. The present study shows that 81 % of African Americans and 46 % of European Americans in the DHS population have the homozygous GG allele associated with high AA levels. In contrast, no African Americans in the present study population with a homozygous TT allele were found, whereas eighteen out of 159 European Americans carried the homozygous TT allele at rs174537. Additionally, as observed by Tanaka et al. (Reference Tanaka, Shen and Abecasis37), there were significant differences in AA and DGLA as well as the AA:DGLA ratio, an estimate of FAD1 activity, between GG, GT and TT in the European American populations. These differences were not seen in the African American population, as the study was not powered to detect such a difference in the small population, which also contained no subjects homozygous for the minor allele in the DHS database. However, a follow-up study has demonstrated comparable (to that in DHS European Americans), highly significant genotypic differences in circulating levels of AA and DGLA and the resultant AA:DGLA ratio in an subset of a larger African American study population (GeneSTAR; Johns Hopkins University, Baltimore, MD, USA) in which the African American sample size was three times that in the present study(Reference Mathias, Sergeant and Ruczinski48).
Third, an analysis of allele frequencies at rs174537 in HapMap populations around the world shows dramatic differences in allele frequencies among populations. For example, populations from Africa have much higher frequencies of the GG alleles along with very low TT allele frequencies. At the opposite end of the spectrum are endogenous populations from the Americas, which exhibit extremely low frequencies of GG alleles and much higher frequencies of the TT alleles. Other populations (a European population from Tuscany, European Americans from the present study and Utah as well as Japanese and Chinese populations) lie in the middle with regard to allele frequencies of this SNP. If indeed the genotype of this SNP (rs174537) correctly predicts ‘efficient converters’ (GG), ‘modest converters’ (GT) and ‘non-converters’ (TT) with regard to Δ5 desaturase (FADS1) enzymic efficiency, then Fig. 3 suggests that a simplistic assumption that wide variations in dietary LA do not alter tissue AA content cannot be made until metabolic studies are carried out in several distinct populations around the world. This is especially important, given that the present paper suggest that the efficiency of LA to AA conversion is probably population dependent.
Finally, numerous studies have demonstrated population differences due to adaptation to pathogens(Reference Haldane49), climate and diet(Reference Luca, Perry and Di Rienzo50). However, some past adaptations (such as salt retention and hypertension) are now maladaptive, and can lead to human disease. We propose that this possibility exists for AA and inflammation. Given the elevated levels of n-6 medium-chain PUFA (12–17 g/d, principally LA(Reference Muskiet, Montmayeur and le Coutre23)) in Western diets, a more efficient capacity to convert medium-chain PUFA to LC-PUFA could promote AA production. This hypothesis is supported by the present study showing that circulating AA in African Americans is on average higher than that in European Americans (Fig. 1), under conditions where genetic backgrounds clearly play a role (Fig. 2). Conditions such as obesity, type 2 diabetes and hypertension are multi-factorial diseases that disproportionately affect African Americans in the USA(44). Yet, only 1–2 % of Africans on the African continent have type 2 diabetes, whereas the incidence is 11–13 % in individuals of African descent in industrialised nations consuming a Western diet(45, 46). There are probably numerous genetic markers and metabolic changes that contribute to these differences, certainly polymorphisms, such as those found in the FADS gene cluster, which could confer increased risk as these populations moved from traditional to Western diets. It is interesting to note that haplotypes of the FADS gene cluster, including variants associated with an elevated AA:LA ratio, are related to both a higher systemic inflammation (as measured by high-sensitivity C-reactive protein) and greater risk of coronary artery disease(Reference Martinelli, Girelli and Malerba31).
For these reasons, recommendations to increase dietary LA levels to 5 to 10 % of dietary energy(Reference Harris, Mozaffarian and Rimm26) by organisations such as the American Heart Association are particularly concerning. These recommendations have come largely as a result of registered clinical trials of mixed n-6+n-3 PUFA diets and diets in which n-6+n-3 PUFA have replaced trans-fatty acids and SFA. Using meta-analyses that took these potential confounders into consideration, Ramsden et al. (Reference Ramsden, Hibbeln and Majchrzak51) have observed a potential risk of LA that was probably missed in previous meta-analyses. Five decades of studies and the clinical impact of inhibitors of AA metabolites or metabolism (non-steroidal anti-inflammatory drugs and leukotriene blockers) support a central role for AA in inflammation. If circulating levels of AA are indeed important, the potential risk of elevating dietary LA would be postulated to differentially affect populations such as African Americans that have a much higher proportion of ‘efficient converters’ of LA to AA, resulting in higher levels of circulating AA.
Acknowledgements
The present study was supported by National Institutes of Health (NIH) grants P50 AT002782 (F. H. C.), R01 Hl637348 (D. W. B.), R01 NS058700 (D. W. B.), F32DK083214 (C. E. H.), RO1 NR08153 (D. M. B.), RO1 DK 071891 (B. I. F.) and M01 RR000052 (D. M. B.). Additional support was received from Wake Forest School of Medicine General Clinical Research Center M01 RR07122 (D. W. B.) and the Wake Forest School of Medicine Center for Public Health Genomics. The authors thank the DHS coordinators, Carrie Smith and Pamela Hicks for their help with obtaining specimens; and Dr R. Mathias for access to study results before publication.
F. H. C. has published books with Rodale and Simon and Schuster and is a founder and consultant to GeneSmart Health, Inc., which may be partially related to his research. These potential conflicts of interest have been disclosed to Wake Forest School of Medicine and to outside sponsors and are institutionally managed. No other authors have a conflict of interest.
C. E. H. performed SNP genotyping and association analyses; M. E. R. selected SNP and designed the genotyping assays; S. S. and P. I. performed fatty acid analyses; H. C. A. performed analyses on the HapMap data; J. T. Z., L. D. C., C. D. L. and D. V. provided statistical and genetic analyses; D. W. B. and B. I. F provided access to DHS data; R. A. M. and F. H. C. generated the hypotheses and designed the experiments; S. S., R. A. M. and F. H. C. prepared the manuscript.





