


There is an urgent need and desire to move away from the currently used trial-and-error or one-size-fits-all approaches in clinical or public health practice, respectively, towards programmes that leverage data on the genomic and phenomic determinants of health/disease to provide more ‘personalised’, ‘stratified’ or ‘precise’ diagnosis, treatment and prevention of common, multifactorial diseases. The flagship UK Biobank programme (www.ukbiobank.ac.uk/), established in 2007, the Genomics England initiative (www.genomicsengland.co.uk/) in 2013, or the more recent US precision medicine initiative project in 2015 (allofus.nih.gov/) all have the common, overarching goal of providing novel insight(s) into disease aetiology and the promise of a new perceived era of ‘better tests, better drugs and above all better, more personalised care to save lives’(1). The reaction to these initiatives has been mixed(Reference Bayer and Galea2, Reference Khoury, Iademarco and Riley3). Most notably, public health experts remain sceptical as they argue an approach that focuses excessively on genes, drugs and disease, whilst ignoring the notion that the major causes of morbidity/mortality in the developed world are potentially preventable – that is, by reducing tobacco or alcohol use, poor diet, physical inactivity and by addressing social differences among populations – remains piecemeal in the case of a majority of non-communicable diseases. They advocate a ‘precision public health’ initiative that moves beyond diagnoses and treatment of individuals to providing the right intervention to the right strata of the population at the right time(Reference Rose4).
Following on from the precision public health-pronged approach, the concept of ‘precision’ nutrition has recently branched out from the ideas underpinning ‘personalised’ nutrition; note that these two terms are often used interchangeably in the literature. Although, there has been little discussion pertaining to the shift from personalised to precision nutrition, from a practical viewpoint the shift broadly describes moving beyond current dietetic-type practice – which bases a dietary plan for a specific disease diagnosis/prognosis, on anthropometric, lifestyle, demographic and psychosocial data – to a setting that also includes genomic data for more targeted and precise adjunct dietary intervention(Reference Lampe, Navarro and Hullar5). This shift within the nutritional sciences has been largely driven by the promises and initial excitement of the genomic medicine revolution, through genome-wide association studies (GWAS), which have been fuelled by the rapid growth in technology offering unprecedented resolution of the human genome at ever decreasing cost. However, putting specific forms of cancer aside, we now know that the genetic variants that have been identified through GWAS explain little of the overall risk or heritability for a majority of the common, multifactorial diseases and are of little practical value for the purposes of ‘prediction’ of individual or group-level outcomes; see Khera et al. (Reference Khera, Chaffin and Aragam6) for an opposing viewpoint. While genetic risk scores (GRS) or polygenic risk scores (PRS) are now being developed in the context of common, multifactorial diseases, their utility in terms of greater predictive performance, or inclusion as part of routine clinical care for better diagnosis, prognosis and monitoring remains largely unknown outside oncology/cancer and warrants formal assessment in pragmatic settings. Furthermore, it is important to state that GRS or PRS have not thus far had any application in micro/macronutrient gene mapping studies. To our knowledge, there are currently no published studies focused on assessing GRS for prediction of micronutrient status. Nevertheless, GRS may play an important role in precision nutrition given that there does seem to be a simpler genetic architecture underpinning population variability in the status of some micronutrients. So, we purport that while personalised nutrition is already commonly used in dietetic practice, and largely without use of genetic information, the move to precision nutrition approaches that advocate the use genetic data is premature. To achieve the full scope of precision nutrition, it is imperative to first review and synthesise the existing evidence – or lack thereof – from large-scale genetic epidemiology studies that have aimed to dissect the complex interplay of diet and genomics before we even contemplate their utility to dissect human health disparities.
To this end, we set out to provide a critical evaluation of findings from GWAS studies of micronutrient status with three overarching aims. First, we aimed to produce a complete catalogue of genetic variants that have been identified through GWAS of micronutrient status. Secondly, we aimed to highlight and critique the principal findings from this catalogue and their utility within clinical or public health practice. Lastly, we aimed to highlight the main gaps that need to be filled to direct future research on the roadmap to precision nutrition. We decided to focus on micronutrients primarily because of their amenability to GWAS studies in terms of function, associations to myriad of common diseases, and their unbiased biochemical assessments.
A catalogue of genes associated with micronutrient status
We used the GWAS catalogue (www.ebi.ac.uk/gwas/) and the National Center for Biotechnology Information (NCBI) to identify all GWAS that have been completed to date in relation to micronutrient disposition. We investigated both biomarkers of micronutrient status and biomarkers of micronutrient function, which provide more insight into cellular metabolism and offer more relevant information regarding the biological and physiological function of the micronutrients. Our findings are summarised in Table 1.
Table 1. Summary of genome-wide association studies (GWAS) that have identified genomic loci associated with micronutrient status in Caucasian adults
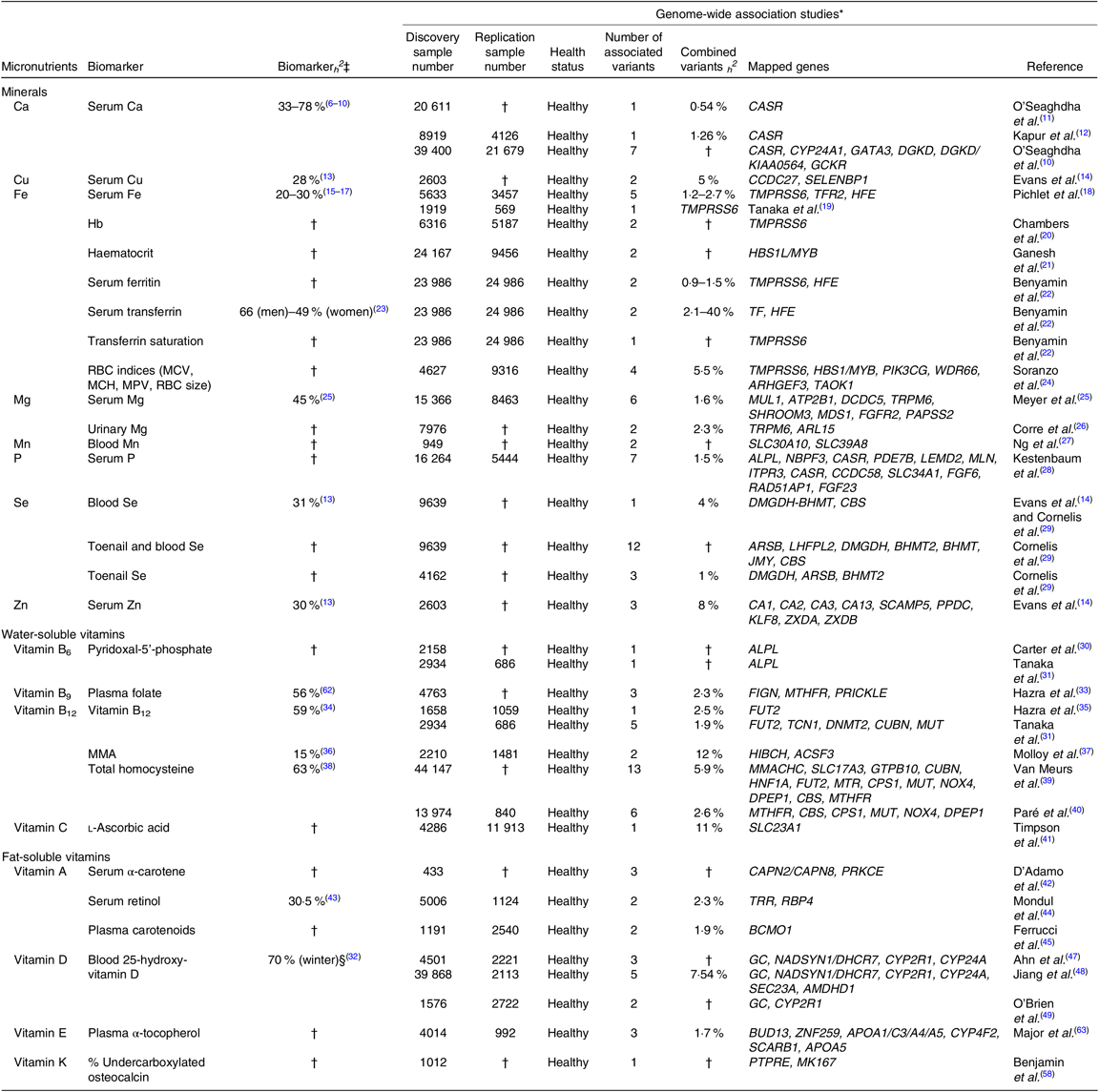
ACSF3, acyl-CoA synthetase family member 3; ALPL, alkaline phosphatase, biomineralized associated; AMDHD1, amidohydrolase domain containing 1; APOA1/C3/A4/A5, apolipoprotein A1/C3/A4/A5; ARHGEF3, rho guanine nucleotide exchange factor 3; ARL15, ADP ribosylation factor like GTPase 15; ARSB, arylsulfatase B; ATP2B1, ATPase plasma membrane Ca2+ transporting 1; BCMO1, β-carotene oxygenase 1; BHMT, betaine-homocysteine methyltransferase; BHMT2, betaine–homocysteine S-methyltransferase 2; BUD13, BUD13 homolog; CA1, carbonic anhydrase 1; CA13, carbonic anhydrase 13; CA2, carbonic anhydrase 2; CA3, carbonic anhydrase 3; CAPN2, calpain 2; CAPN8, calpain 8; CASR, Ca sensing receptor; CBS, cystathionine-β-synthase; CCDC58, coiled-coil domain containing 58; CCDC27, coiled-coil domain containing 27; CPS1, carbamoyl-phosphate synthase 1; CUBN, cubilin; CYP24A, cytochrome P450 family 24 subfamily A member; CYP24A1, cytochrome P450 family 24 subfamily A member 1; CYP2R1, cytochrome P450 family 2 subfamily R member 1; CYP4F2, cytochrome P450 family 4 subfamily F member 2; DCDC5, double cortin domain containing 5; DGKD, diacylglycerol kinase delta; DHCR7, 7-dehydrocholesterol reductase; DMGDH, dimethylglycine dehydrogenase; DNMT2, DNA methyltransferase-2; DPEP1, dipeptidase 1; FGF23, fibroblast growth factor 23; FGF6, fibroblast growth factor 6; FGFR2, fibroblast growth factor receptor 2; FIGN, Fidgetin; FUT2, fucosyltransferase 2; GATA3, GATA binding protein 3; GC, group-specific component; GCKR, glucokinase regulatory protein; GTPB10, GTP binding protein 10; HBS1L, HBS1 like translational GTPase; HFE, homeostatic Fe regulator; HIBCH, 3-hydroxyisobutyryl-CoA hydrolase; HNF1A, HNF1 homeobox A; ITPR3, inositol 1,4,5-trisphosphate receptor type 3; JMY, junction mediating and regulatory protein, P53 cofactor; KIAA0564, Von Willebrand factor A domain containing 8; KLF8, kruppel like factor 8; LEMD2, LEM domain containing 2; LHFPL2, LHFPL tetraspan subfamily member 2; MCH, mean corpuscular Hb; MCV, mean corpuscular volume; MK167, marker of proliferation Ki-67; MLN, motilin; MMA, methylmalonic acid; MMACHC, metabolism of cobalamin associated C; MPV, mean platelet volume; MTHFR, methylenetetrahydrofolate reductase; MTR, 5-methyltetrahydrofolate-homocysteine methyltransferase; MUL1, mitochondrial E3 ubiquitin protein ligase 1; MUT, methylmalonyl-CoA mutase; MYB, MYB proto-oncogene; NADSYN1, NAD synthetase 1; NBPF3, NBPF member 3; NOX4, NADPH oxidase 4; SLC23A1, solute carrier family 23 member 1; PAPSS2, 30-phosphoadenosine 50-phosphosulfate synthase 2; PDE7B, phosphodiesterase 7B; PIK3CG, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit Y; PPDC, prepilin peptidase-dependent protein C; PRICKLE, prickle planar cell polarity protein; PRKCE, protein kinase C epsilon; PTPRE, protein tyrosine phosphatase, receptor type E; RAD51AP1, RAD51 associated protein 1; RBC, erythrocyte; RBP4, retinol binding protein 4; SCAMP, secretory carrier membrane protein; SCAMP5, secretory carrier membrane protein 5; SCARB1, scavenger receptor class B member 1; SEC23A, Sec23 homolog A, coat complex II component; SELENBP1, Se binding protein 1; SHROOM3, shroom family member 3; MDS1, myelodysplasia syndrome 1 protein; SLC17A3, solute carrier family 17 member 3; SLC30A10, solute carrier family 30 member 10; SLC34A1, solute carrier family 34 member 1; SLC39A8, solute carrier family 39 member 8; TAOK1, TAO kinase 1; TCN1, transcobalamin 1; TF, transferrin; TFR2, transferrin receptor 2; TMPRSS6, transmembrane serine protease 6; TRPM6, transient receptor potential cation channel subfamily M member 6; TRR, transfer RNA arginine; WDR66, WD repeat domain 66; ZNF259, Zn finger protein 1; ZXDA, Zn finger X-linked duplicated A; ZXDB, Zn finger X-linked duplicated B.
* Results presented in this table are genome-wide significant (P < 10−7), and/or significant at P < 0·05 level in replicated, independent Caucasian cohorts.
† Indicates that no results were found.
‡ All heritability results are from twin studies, except serum retinol (family study).
§ Serum 25-hydroxy-vitamin D levels were found to be highly heritable during winter months only, and not heritable at all during summer months(Reference Karohl, Su and Kumari32).
To our surprise, we found that all major micronutrients – including, minerals, fat- and water-soluble vitamins – have been studied through GWAS, and that a varied number of loci associated with status of each micronutrient have been identified. Notably, a majority of the micronutrient-related heritability or GWAS have been carried out using healthy, Caucasian populations, which is why our summary table is limited to these criteria.
Heritability of micronutrient status
A number of important summary statistics can be extracted from Table 1. Estimates of heritability (h2) ranged from 23 % for serum Fe status up to 70 % for 25-hydroxy-vitamin D. In general, mineral statuses were the least heritable (mean h2 = 35 %) followed by water (mean h2 = 48 %) – and fat-soluble (mean h2 = 50 %) vitamin status. We failed to identify a reported h2 for a sizable number of micronutrients. In fact, of the twenty-two micronutrients reported in Table 1, only ten had a reported h2 value and they were predominantly based on twin rather than family studies. There was very good coverage of the minerals with only two – Mn and P – out of the eight minerals not having a reported h2 value. Ca status was by far the most heritable mineral at h2 = 61 % whilst all other minerals exhibited low heritability values. Of the water- or fat-soluble vitamins, for example, only vitamins A (serum retinol measurements), B9 (erythrocyte folate), B12 and D have reported h2 values. Perhaps not surprisingly, in the case of 25-hydroxy-vitamin D, only the winter measurements were heritable and status of 25-hydroxy-vitamin D was not heritable at all during summer months(Reference Karohl, Su and Kumari32). It is important to note that we did not identify any heritability studies in non-Caucasian populations.
Genome-wide association studies of micronutrient status
We identified twenty-two micronutrients that had at least one GWAS reported and of the twenty-two all identified at least one significant (P < 10−7 in discovery and significant at P < 0·05 level in replicated, independent cohorts) associated variant. Those micronutrients missing from the GWAS catalogue were B, Co, Cl, Cr, iodine, Mo, K, Na, vitamins B1, B2, B3, B7 and choline.
The proportion of variance explained by the identified variants – individually or collectively – ranged from <1 % in the case of serum ferritin to 11 % for vitamin C status (l-ascorbic acid concentrations) and in general the majority of the variants explain little of the overall heritability in status for each micronutrient although there are notable exceptions. For example, a single common single nucleotide variant in the genes SLC23A1 and HIBCH explains 11 and 12 % of the inter-individual variability in l-ascorbic acid and methylmalonic acid (MMA – a functional biomarker of vitamin B12 status), respectively(Reference Timpson, Forouhi and Brion41, Reference Molloy, Pangilinan and Mills46). Moreover, in the case of minerals generally, often a small number of variants explain a sizable proportion of the heritability; on average, 14 % (range: 2–28 %) of the heritability in mineral status is explained by the variant. The situation is far less clear for water-/fat-soluble vitamins mainly due to unavailability of adequate data for a majority of these micronutrients. Furthermore, we could not identify reported estimates of the variability of variants identified in a number of GWAS, including those of Mn (the only mineral to not provide this estimate), and vitamins B6, pro-vitamin A serum α-carotene, vitamin D and vitamin K.
Lessons from heritability and genome-wide association studies of micronutrient status
A number of observations and inferences can be made about the available data but four points warrant a particular mention. First, it is important to state that certain aspects of the data from Table 1 stand in direct contrast to findings from GWAS of common traits/diseases where often a very large number of loci explain a small fraction of the inter-individual variation or trait heritabilities. Second, we found a dearth of (discovery) studies undertaken in other ethnic groupings, although a small number of replication studies were undertaken in East Asian and African-American cohorts(Reference Kim, Kwangsik and Ramanana50–Reference Li, Lange and Duan54). Third, there seems to be a general lack of utility provided by the identified genetic variants for prediction of individual and/or group-level status although GWAS have provided some novel insight into mechanisms that underpin inter-individual variability in micronutrient status. This is exemplified in the recent study by Molloy et al. (Reference Molloy, Pangilinan and Mills46). Their replicated findings showed that serum MMA levels were unexpectedly largely influenced by a cobalamin-independent pathway involved in valine-catabolism mediated by the HIBCH gene product. Elevated plasma MMA concentrations are clinically used to diagnose individuals with inborn errors of metabolism or with vitamin B12 deficiency (i.e. the elderly). The discovery of this SNP (1) questions the clinical utility of circulating MMA assessment in diagnosing cobalamin deficiency, and (2) may be relevant to study and consider in individuals possessing the variant who are in states of acute disease. Finally, we think the data clearly highlight the current gaps in knowledge and the significant challenges ahead that need to be addressed before we entertain the scope of precision nutritional approaches that leverage genetic information in either clinical or public health settings.
Gaps in knowledge and challenges ahead
In our opinion, there are three overarching challenges that warrant particular attention. First and foremost, most of the data presented in Table 1 are derived from studies based on supposedly ‘healthy’ variability in status. Furthermore, there is a dearth of heritability or GWAS studies that aim to partition the genetic basis of ‘functional’ micronutrient deficiency or studies that have evaluated the role of the identified loci in Table 1 in increasing the risk of functional deficiency. Secondly, we identified only one GWAS of (differential) ‘response’ to micronutrient intervention in either health or disease, although there are some candidate gene/polymorphism studies. This is a particularly important issue as currently the underlying genetic architecture of micronutrient ‘status’ and ‘response’ to intervention remains totally unknown. Indeed, there is little point in identifying strata of the population at elevated, genetically-driven risk of functional deficiency if they will not benefit from supplementation (i.e. due to lack of beneficial response) as a result of an alternative genetic cue. Thirdly, an obvious question remains as to the portability of findings from GWAS across populations, which could hinder the translation of findings into relevant recommendations/applications in different ethnic groups. As shown in Table 1, the current data stem overwhelmingly from Caucasian discovery cohorts motivating future studies that aim to test and identify population-specific and trans-ethnic micronutrient-associated genetic loci. Although questions about the complexity of gene–nutrient interactions and their role in diseases with environmental causes have been previously discussed, it is nonetheless vital to explore in more depth the adaptability of molecular processes modelled by different environments and contexts.
Unravelling the genetic basis of functional micronutrient deficiency
As outlined in Table 1, the application of GWAS has allowed us to identify genetic loci associated with static and, when available, functional biomarkers of micronutrient status in healthy subjects. We would like to highlight a general lack of adequate functional biomarker(s) of micronutrient status that are sensitive in distinguishing between healthy variability, insufficiency and overt or severe deficiency, particularly at different levels of dietary exposure. Notably, there seems to be a gulf of either unavailability of a functional biomarker per se, as is the case for Cu and Zn, to unavailability of biomarkers for assessing status in replete, healthy subjects.
The lack of adequate functional biomarkers thus represents a major Achilles heel when trying to unravel the contribution of genetic variation to the full spectrum of micronutrient status. This has been discussed in depth by Combs in the case of Se status but the topic warrants a more general discussion across all the micronutrients, which is beyond the scope of the current commentary(Reference Combs55). Perhaps a good case example that highlights the complexities associated with genetic studies of static and functional biomarkers of micronutrient status is represented by a recent GWAS of vitamin B12 status. For instance, there are now a number of GWAS studies of serum vitamin B12 levels as well as a recent GWAS of serum MMA – a functional early and specific indicator of (mitochondrial) vitamin B12 status/deficiency. As can be seen from Table 1, there are five independent loci that have been shown to be significantly associated with serum vitamin B12 concentrations in healthy adults, including common genetic variants in genes FUT2, TCN1, DNMT2, CUBN and MUT which collectively explain approximately 3 % of the heritability in serum vitamin B12 levels. A notable finding from the recent GWAS of MMA (2016) was the discovery of genetic variants in the gene 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) and acyl-CoA synthetase family member 3 (ACSF3) genes that collectively explain approximately 12 % of the variance in MMA concentration in healthy adults. We have recently shown that the heritability for normal MMA levels is approximately 15 % which means that >80 % of the heritable variation in MMA is accounted for by variation at these two genes (under review). What is important about this finding is that genetic variation at neither of these two genes is associated with serum vitamin B12 concentrations and none of the five genes associated with serum vitamin B12 concentrations are associated with MMA concentration in healthy adults. This highlights the difficulties associated with using biomarkers of deficiency to study health, given previous findings from randomised controlled trials and systematic studies that have shown that plasma vitamin B12 concentrations, MMA and total homocysteine are effective biomarkers of vitamin B12 status(Reference Hoey, Strain and McNulty56).
Our search of both the published literature and the GWAS catalogue, in fact, highlighted a dearth of genetic epidemiology research aimed at unravelling the relative importance and contribution of genetic and environmental factors to the cause of functional micronutrient deficiency. Although inadequate exposure is often cited as the most important cause of deficiency, there are a number of potential factors that may govern micronutrient bioavailability – including absorption, distribution or transport, metabolism, elimination/excretion (ADME) that may manifest over time as micronutrient deficiency. These include negative nutrient–drug interactions(Reference Péter, Navis and de Borst57), physiological changes through ageing, occurrence of disease or pathophysiological changes that occur predisease diagnosis, dietary intervention and genetic factors cross-cutting all these processes that can either affect ADME or increase the risk of the processes affecting aberrant ADME processes.
We note that out of all the micronutrients reported in Table 1, only two, Fe (serum ferritin) and vitamin K (percentage decarboxylated osteocalcin), were studied in deficient subjects(Reference Benjamin, Dupuis and Larson58, Reference Mclaren, Garner and Constantine59) and, to our knowledge, there are currently no reported heritability studies of functional micronutrient deficiency per se. Although not directly comparable, we note that there were no overlaps between findings of GWAS of deficiency in Fe or vitamin K and healthy status. As shown in Table 1, genome-wide significant loci were mapped to the IGL and to PTRPE and MK167 loci in the case of Fe and vitamin K deficiency, respectively. None of these loci were shown to be significant in GWAS of Fe or vitamin K in non-deficient, healthy subjects(Reference Benjamin, Dupuis and Larson58). This may indeed be a function of the GWAS approach and the difficulty in detecting associated loci at genome-wide significance(Reference Marini, Yang and Asrani60). On this point, we would advocate the use of a combination of genetic approaches (candidate polymorphism, gene and pathway) and would like to draw attention to two alternative gene mapping efforts that we believe warrant particular attention for precision nutrition approaches. First, a recent case finding from the 100 000 genomes project illustrates the use of large-scale sequencing approaches for precision nutrition. A newborn with fluctuating neurology and immunodeficiency was one of the participants of the 100 000 genomes project. Although sadly the child passed away, deep sequencing led to the identification of mutations in the TCN2 gene, which lead to defective vitamin B12 transport. Case reports had suggested high doses of vitamin B12 might overcome this and indeed it seems this has helped a second child in the family. The scope of this finding is best illustrated by the study of Marini et al., which has shown that the effects of some of the functional mutations in the MTHFR gene, which impact activity of the methyl transferase enzyme, can be remedied through folic acid supplementation(Reference Marini, Gin and Ziegle61), further supporting the idea that the identification of polymorphisms that have subtle effects on functional micronutrient status could provide us with further insights for the remedial of dysfunctional enzymes, and could potentially be of use in providing corrective measures to allele carriers on a larger scale.
Deciphering the genetic basis of variable response to dietary micronutrient intervention/exposure
A major gap in the roadmap to precision nutrition is the lack of knowledge, appreciation and, worryingly, dearth of research into unravelling the basis and relative contribution of acquired and/or inherited factors to the observed variability in response to dietary micronutrient exposure or intervention. Consequently, the underlying genetic basis, and similarities and/or differences in the genetic basis of ‘status’ and ‘response’ to intervention remain largely unexplored.
There are only a handful of studies that have attempted to (i) partition the relative importance of genetic and environmental factors in response to dietary interventions; or (ii) identify genetic variants associated with response to dietary intervention. To our knowledge, only one classical twin study (n 101 twin pairs), using a ‘before and after’ intervention trial of 400 µg of folic acid per d for a period of 6 weeks, has calculated heritabilities of plasma folate status and total homocysteine (tHcy) concentrations at baseline, post-dose and response to intervention. Furthermore, they tested whether the MTHFR C677T polymorphism, which is known to be associated with steady-state – that is, baseline levels – plasma folate or tHcy concentrations, is also associated with differential response to folic acid intervention. The results showed that both steady-state levels (h2 = 59 %) and ‘response’ to folate intervention were both highly heritable (h2 = 64 %) with 22 % of the cohort failing to respond folate supplementation – that is, no change in tHcy – although most participants had up to a 3-fold increase in folate status. Notably, the MTHFR C677T polymorphism was not associated with response to intervention although it showed association with baseline levels of tHcy(Reference Cotlarciuc, Andrew and Dew62). In support of this observation, two separate GWAS on the Beta-Carotene Cancer Prevention Study cohort (2011 and 2012) sought to identify common genetic variants associated with both vitamin E status(Reference Major, Yu and Wheeler63) and serological response to vitamin E supplementation(Reference Major, Yu and Chung64). In line with findings from Cotlarciuc et al. (Reference Cotlarciuc, Andrew and Dew62), given power and design constraints, the genetic architecture of status and response in the case of vitamin E seem to be largely discordant with no genes and variants therein associated with both outcomes. The present study also highlights a lack of clarity as to what ‘response’ would entail. For example, in the studies by Major et al. (Reference Major, Yu and Wheeler63, Reference Major, Yu and Chung64) a lack of serological response to vitamin E supplementation could either mean that the vitamin E was not absorbed or that it may simply be cleared from the circulation very quickly in some people as a result of genetic factors.
Finally, Mao et al. (Reference Mao, Bath and Vanderlelie65) carried out a candidate polymorphism association study, using the Selenium in Pregnancy Intervention (SPRINT) study of 227 pregnant women, to test for associations between common genetic variation in four genes (DMGDH, SEPP1, GPx1, and GPx4), previously associated with Se status in non-pregnant populations, to Se status in the first trimester of pregnancy, its longitudinal change across the three trimesters, and response to Se supplementation of 60 µg per d. Briefly, the study replicated the known association between the common, candidate polymorphism in the gene DMGDH and Se status (based on both toenail and whole-blood) explaining approximately 2 % of the variance in whole-blood Se concentration at 12 weeks of gestation. In unsupplemented women, the candidate polymorphism in the SEPP1 gene was significantly (P ≤ 0·005) associated with the longitudinal change in whole-blood Se levels from 12 to 20 through to 35 weeks of gestation, explaining 8 % of the variance in this healthy change in Se status across the three trimesters. The same polymorphism in the SEPP1 gene was associated with adequate response to Se supplementation, as measured by the change in GPx3 activity from 12 to 35 weeks of gestation, explaining >5 % of the variance in response to intervention(Reference Mao, Bath and Vanderlelie65). The results from the present study highlight that given an appropriate study design and power, genetic variants can explain a sizable fraction of the variability of healthy change in Se status throughout pregnancy as well as beneficial response to intervention in insufficient/deficient pregnant mothers. Importantly, these results highlight the inherent complexities associated with interpretation of gene/nutrient relationships given different contexts – pregnancy, old age and disease status – and motivate systematic studies, using novel designs in a myriad of settings, aimed at unravelling the genetic basis of response due to a change in environmental context or to dietary interventions and the identification of subpopulations of hyper, normal and hypo responders to intervention.
Identifying trans-ethnic and ethnic-specific genetic effects
To date, all micronutrient biomarker status heritability studies have been done on Caucasian population groups, and very few GWAS have investigated genetic association to micronutrient status in other ethnicities. In fact, merely two association studies have been done on vitamin B12 status in Asian populations(Reference Lin, Lu and Gao52, Reference Nongmaithem, Joglekar and Krishnaveni53), and one on Fe status in African Americans(Reference Li, Lange and Duan54). Consequently, there is a major gap and skew in knowledge with regard to understanding the genetic basis of micronutrient status variability in different ethnicities, potentially tempering the portability of GWAS findings within and across populations of different ancestry and the translatability of the latter into relevant public health application.
Several studies have demonstrated the importance of trans-ethnic replication in the field of personalised medicine. For instance, the benefits of trans-ethnic GWAS with regard to both discovery and characterisation of complex trait loci have been exemplified in a trans-ancestry genome-wide meta-analysis, in which trans-ethnic association analysis allowed the identification of additional type 2 diabetes susceptibility loci, thus extending knowledge into the genetic architecture of the disease(Reference Mahajan, Go and Zhang66). Additionally, a study that ran a comprehensive survey of GWAS replicability across twenty-eight diseases found that some SNP–disease associations failed to replicate in East Asians, and that these SNP were mapped to genomic regions where linkage disequilibrium significantly varies between populations(Reference Marigorta and Navarro67). Trans-ethnic mapping is a potentially powerful tool in identifying genetic variants underlying micronutrient status variability, and subsequently improving public health application/intervention for the purposes of disease prevention. More trans-ethnic GWAS are needed to evaluate the genetic architecture differences in linkage disequilibrium patterns that may be driving genetic associations, and to gain a deeper understanding of the role of identified genetic variants in the complex genetic architecture of micronutrient status variability(Reference Li and Keating68).
Findings from our literature search have also highlighted the existence of population/ethnic-specific genetic variants. Most notably, the vitamin B12-associated FUT6 gene was mapped in Asians, and the Fe-associated MAF and HDGFL1 genes were only mapped in African Americans, but not in Caucasians. In contrast, the TF gene, encoding the transferrin protein, was identified in both Caucasians and African Americans. Identifying pan-ethnic and ethnic-specific variants in a wider range of ethnicities therefore remains a major priority in the roadmap to precision nutrition, as they (1) could potentially provide us with the know-how to ethnically stratify populations, and would be a stepping stone in gaining a deeper understanding of the gene–nutrient interactions attributed to such stratification, and (2) are essential for the portability of GWAS results, and for making distinctions between genome-wide associations driven by population-specific variants in multi-ethnic studies.
Context-dependency of molecular behaviour
While the interest in understanding the links between the genome, environment and complex disease risk is growing, questions regarding the importance of unravelling the gene–nutrient interactions for the purpose of personalised or precision nutrition have been brought forward(Reference Khoury and Evans69, Reference Mozaffarian70). In fact, it has been suggested that such efforts may not be needed, and that the use of biochemical and/or anthropometric indices may be enough in formulating personalised dietary interventions, as these are environmental risk factors known to influence disease risk – namely CVD risk(Reference Mozaffarian70). To that, we respond that investigating gene–nutrient interactions should remain a priority for the refinement of dietary interventions and the management of disease risk, as robust evidence now suggests that genetic variants (1) are able to modify the response to interventions(Reference Heianza and Qi71), and (2) can trigger adverse health outcomes when exposed to high-risk environmental factors(Reference Heianza and Qi71), a concept we term ‘context-dependency of molecular behaviour’. Additionally, several studies have shown that gene polymorphisms respond to nutrient interaction, affecting biochemical markers of metabolic and CVD(Reference Tai, Corella and Demissie72–Reference Zheng, Parnell and Smith74) and obesity(Reference Heianza and Qi71). However, how these polymorphisms interact with nutrients in different contexts of health, disease, drug interventions and across ethnicities has not been investigated to date, and is needed for consideration in future research.
Further to investigating the molecular behaviour of micronutrient–gene interactions in the abovementioned scenarios, a longer-term goal would be to explore their molecular individuality, aiming to understand how molecular systems change with respect to different environmental contexts. A paper by Thompson et al. (Reference Thompson, Kuttab-Boulos and Witonsky75) showed unusual geographic dependency of the CYP3A*3 polymorphism – a gene encoding monooxygenases that catalyse reactions involved in drug metabolism and steroid synthesis – frequency with relative distance from the equator. Interestingly, CYP3A*3 frequency significantly correlated with that of another variant, M235T, known for its implication in hypertension. Findings also suggested that both variants exhibited strong frequency variations across populations and were significantly influenced by environmental factors correlated with latitude(Reference Thompson, Kuttab-Boulos and Witonsky75). Such shared selective pressure calls for further investigation of molecular behaviour in response to a range of environmental stimuli. Identifying the adaptability of these molecular behaviours in response to both short-term and long-term environmental exposures (e.g. pregnancy, healthy ageing, cross-culturally or different living arrangements) would allow further sub-stratification of population groups, and would open new avenues for a more ‘targeted’ and ‘precise’ public health approach.
Conclusions
Advances in technology, powerful biobanks and implementation of large-scale ‘omics’ initiatives and genomic medicine have quickly transcribed a new era of nutritional risk management based on the concept of precision nutrition, promising the implementation of precise preventative measures to thwart the burden of non-communicable disease in the population. However, if the full scope of precision nutrition is ever to be realised, the rigour and direction of the research needs to be evaluated and re-focused. Our overarching conclusion is that, while GWAS have delivered some significant new mechanistic insights, on their own they will not deliver on the vision of precision nutrition. We believe that this type of analysis highlights the need to pause to consider and refine experimental strategies. In the absence of knowledge about how and why individuals respond so differently to dietary changes and intervention, we believe that there is currently insufficient molecular and physiological knowledge to be able to provide robust guidance on how best to resolve micronutrient deficiencies in an individualised or population-based manner. It is important to use the results from Table 1 as incentives for (i) discovery of functional, robust biomarkers of status; (ii) investigating the underlying mechanisms of the established associations, as this holds better promise in advancing precision nutrition than predicting health/disease/status outcomes does; and (iii) identifying genetic variants associated with functional deficiency, nutritional disease, response to supplementation, trans-ethnic and ethnic-specific effects. Furthermore, gaining a deeper understanding of the behavioural changes of molecular systems in different environmental contexts (e.g. pregnancy, old age, disease) remain important priorities in shifting from the current one-size-fits-all and trial-and-error dietary public health approaches to a more precise population-based approach. Finally, open acknowledgment of how little we currently know per se. For example, who could argue with the notion that the microbiome and the vast genetic variability therein plays major role in governing inter-individual variability in status and response to dietary exposure? There is a growing literature on this exciting area of research but more work is needed to delineate the role of the microbiome and assess their utility in practice(Reference Zmora, Zeevi and Korem76–Reference Zeevi, Korem and Zmora79).
Acknowledgements
Conception and design were by K. R. A.; M.-J. D. and K. R. A. carried out data acquisition and interpretation. M.-J. D., R. E. and K. R. A. all contributed to drafting the manuscript and revision for intellectual content. All authors read and approved the final manuscript.
There were no conflicts of interest.

