


This report describes the genetic evaluation of a girl with bilateral Duane retraction syndrome (DRS), cleft palate, and mildly reduced hearing who was found by high-resolution array–comparative genomic hybridization (array CGH) to have a chromosomal anomaly involving a single gene known to be involved in muscle development.
The study was approved by the Institutional Review Board of the College of Medicine at King Saud University, Riyadh, Saudi Arabia, and informed consent was obtained. Patients’ medical records were reviewed, including multiple examinations by both the Ophthalmology and Otolaryngology Departments at King Abdulaziz University Hospital over a period of almost 15 years. Data extracted included family history, complete ophthalmologic and neurologic examinations, laboratory results, and neuroimaging. Genes associated with syndromic DRS (SALL4, CHN1, TUBB3, HOXA1, and KIF21A) were sequenced; the complete coding regions of the SALL4, CHN1, TUBB3, and HOXA1 genes and exons 8, 20, and 21 considered hotspot for mutations in the KIF21A gene were sequenced according to protocols described previously.Reference Abu-Amero, Bosley, Kondkar, Oystreck and Khan 1
The Affymetrix Cytogenetics Whole-Genome 2.7M array (Affymetrix Inc., Santa Clara, CA, USA) was used to detect known and novel chromosomal aberrations across the entire genome. The array CGH assay was performed according to the manufacturer’s instructions as detailed elsewhere.Reference Abu-Amero, Bosley, Kondkar, Oystreck and Khan 1 Data were analyzed using the Affymetrix Chromosome Analysis Suite, v1.2, software. In the absence of internationally recognized criteria for analysis of high-resolution array CGH results, we devised preliminary criteria for a copy number variant (CNV) to be considered potentially pathologic, including: (1) it was not reported in the Database of Genomic Variants (DGV; http://projects.tcag.ca/variation/) among normal controls; (2) it was not present in 150 healthy controls of similar ethnicity; (3) it included an area of the genome encompassing one or more functional genes; and (4) it segregated with the phenotype and was not present in unaffected family members. The threshold for gain or loss was adjusted to 10 kb. We used the National Center for Biotechnology Information Human Genome Assembly Build 35.
The proband was a 16-year-old girl with bilateral type 3 DRS. Her parents were first cousins. Her father was reportedly asymptomatic, but her mother had congenital strabismus that was treated with strabismus surgery during childhood. Three of her mother’s seven siblings reportedly had congenital strabismus as well. These individuals could not be examined, but none had features of DRS by report. The proband had four unaffected siblings and a brother with congenital left superior oblique palsy.
She was the product of a normal pregnancy and delivery, but was born with a cleft palate that was successfully repaired at the age of 1 year. Her hearing was modestly reduced bilaterally with flat tympanograms, but she did not require hearing aids. She achieved normal developmental milestones, although her speech from early childhood through her teenage years was modestly abnormal with poorly formed words and multiple word substitutions, possibly as the result of her hearing difficulties. Cognitive function was grossly normal, and she did not display any autistic features.
At age 16 years, the proband’s visual acuity measured 20/30 OU with excellent color vision and normal appearing optic discs and posterior poles bilaterally. She had a small esotropia and right hypertropia in forced primary position and, in general, assumed a small face turn and head tilt to the left. She had bilateral DRS type 3 with almost complete absence of abduction and modest deficits of adduction associated with marked retraction of each eye on attempted adduction (Figure 1). An upshoot of the nonfixing adducting eye could be elicited during attempted horizontal gaze if the eyes were slightly above midline. Additionally, a downshoot of the left eye would occur if attempting right gaze slightly below the horizontal midline. Convergence was relatively poor. A magnetic resonance imaging scan of the brain and orbits performed at age 11 years was entirely normal, including extraocular muscles (EOMs), except that the abducens nerves were not well seen, possibly because of motion artifact in the CSF anterior to pons.
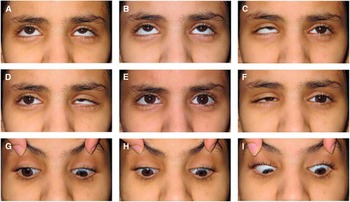
Figure 1 Ocular motility in proband. External photos in nine positions of gaze (A-I). Primary position (E) shows small esotropia and hypertropia of the nonfixing right eye. Right (D) and left gaze (F) show marked deficits of adduction, moderate deficits of adduction, and obvious narrowing of the palpebral fissure secondary to globe retraction of either adducting eye. Elevation of the adducting eye could be readily demonstrable if eyes were slightly above midline during attempted horizontal gaze (D); otherwise, upshoot would not occur if eyes remained along the midline (E). Vertical gaze was full (B and H) with persisting deficits of horizontal movement during attempted oblique positions of gaze (A, C, G, and H).
No sequence variations were detected in the screened regions of SALL4, CHN1, HOXA1, TUBB3, and KIF21A genes. Array CGH documented a 12-kb deletion in chromosome X extending from 32,568,156 to 32,580,298 and encompassing part of only the dystrophin gene (Gene Symbol DMD; NC_000023.11) extending from exon 14 to intron 16. The deletion involves repeat 2 and repeat 3 encoding the central rod domain of the DMD gene that comprises of 24 spectrin-like repeats folded in triple coiled-coil structure. The DMD gene reading frame checker at the Leiden Muscular Dystrophy database (www.dmd.nl), which predicts the effect of exon deletion/duplication on the reading frame indicated that deleting the exon 14 through exon 15 leads to an in-frame deletion. Further analysis using the eDystrophin database (http://edystrophin.genouest.org/), which predicts the consequences of the in-frame mutations at the protein level, indicated that the deletion of exons 14-15 (c.1603-?_1812+?del; p.Val535_Ala640del) partially affects the lipid-binding domain 1 of the DMD protein and may not allow the protein reconstituting a triple-coiled coil at the new junction of the two sides of the deletion, leading to hybrid or fractional repeat, and the filamentous structure may not be maintained. The copy number state was equal to 1, indicating that this deletion was likely to be heterozygous. The confidence value calculated by the Chromosome Analysis Suite software was 88%, with a marker count of 16 spanning the deleted area. This deletion was absent in the proband’s mother and father and was therefore likely to be de novo. It was not present in the DGV or in 150 unrelated healthy individuals of similar ethnicity.
The patient described here is a young woman with bilateral syndromic DRS. She also had a cleft palate and moderately poor hearing bilaterally. DRS, cleft palate, and partial deafness occur in Wildervanck syndrome, but she did not have the Klippel-Feil anomaly. She did not exhibit any of the additional features of recognized monogenic syndromes associated with DRS,Reference Bosley, Abu-Amero and Oystreck 2 and did not have mutations in any of the genes (SALL4, CHN1, HOXA1, TUBB3, or KIF21A) known to cause DRS or ocular motility problems similar to DRS. Array CGH revealed a small heterozygous deletion of only the dystrophin gene on the X chromosome that was not present in her parents, the DGV, or 150 ethnically matched normal controls.
This is the first report to describe the presence of a heterozygous mutation or deletion of the dystrophin gene in a female patient with DRS. However, the coincidence of DRS with dystrophin mutations in the setting of males with Duchenne muscular dystrophy has been described three times previously, including a recent report by our group describing a boy with bilateral DRS, unequivocal DMD, and a small X chromosome duplication involving exons 3 and 4 of the dystrophin gene.Reference Bosley, Salih and Alkhalidi 3 These three patients imply a possible association between dystrophin gene changes and DRS that has not yet been evaluated in a larger group of patients with nonsyndromic or syndromic DRS. Interestingly, single nucleotide polymorphisms in the dystrophin gene have been found in association with oral clefts,Reference Patel, Beaty, Ruczinski, Murray, Marazita and Munger 4 creating a potential association between mutated dystrophin and facial clefts that are at times also associated with congenital abnormalities of ocular alignment and motility.
Mutated dystrophin protein is present in EOM fibers of patients with DMD and animal models of DMD,Reference Matsumura, Ervasti, Ohlendieck, Kahl and Campbell 5 raising the possibility that dystrophin mutations could affect EOM development at times. In general, there are no signs of muscle fiber degeneration, connective tissue accumulation, or central nuclei in EOM of DMD animal models, and eye movements in DMD patients are typically normal except for somewhat slowed saccades. Although EOMs are very fast and active, the loads they work against are comparatively small, possibly making them less susceptible to mechanical injury that dystrophin may help protect against. There is also a previous report of a congenital ocular motility abnormality with globe retraction similar to DRS associated with the gene XIRP2 involved predominantly in muscle development, implying that the primary genetic abnormality in some patients with congenital ocular motility abnormalities may be related to muscle development.Reference Abu-Amero, Bosley, Kondkar, Oystreck and Khan 1
Nevertheless, the presence of DRS in the patient described in this study with a heterozygous dystrophin mutation and in three other patients with DMD raises the possibility that the frequency of DRS may be affected by dystrophin mutations. In DMD, functional dystrophin is also missing in postsynaptic regions of the cerebellum and cerebral cortex, and central nervous system involvement in DMD is confirmed by the presence of cognitive deficits and increased cortical excitability. The presence of dystrophin mutations in either neurologic or ocular muscle tissue (or both) might play a role in this patient. In particular, dystrophin in the embryonic lateral rectus muscle could be a factor at times in the process of establishing normal development of EOM function and innervation, and dystrophin mutations may at times disturb that process.
This report has several limitations. Both the proband’s cleft palate and her family history of strabismus offer alternative (possibly partial) explanations for syndromic DRS, although up to 20% of patients with DRS have a family history of strabismus. This report describes only one female patient with DRS associated with a dystrophin mutation. Even though three previous male patients with DMD have been reported to have DRS, the potential role of dystrophin as a factor in the occurrence of DRS cannot be fully addressed until more patients are identified with similar CNVs and/or until more patients with nonsyndromic and syndromic DRS are evaluated for possible dystrophin single nucleotide polymorphisms or mutations. Nevertheless, it is valuable to consider the possibility that the original congenital fibrosis of extraocular muscles (CFEOM) concept of a primary muscle developmental abnormality causing congenital ocular motility problems may not have been completely incorrect, even after two decades of identifying only neurogenic causes of the congenital cranial dysinnervation disorder (CCDDs).Reference Bosley, Abu-Amero and Oystreck 2 In conclusion, this patient raises the possibility that dystrophin may on occasion be a factor in the development of DRS and certainly offers a reason to continue to be vigilant for both myopathic and neurogenic genetic factors involved in DRS and other CCDDs.
Acknowledgments
We are indebted to the Glaucoma Research Chair at King Saud University, Riyadh, Saudi Arabia, for allowing us to use the laboratory facilities. This project was supported by the Kingdom of Saudi Arabia National Program for Science and Technology Grant #12-MED2621-02, King Saud University, Riyadh, Saudi Arabia.
Disclosures
The authors have no disclosures to report.

