



Introduction
Nuclear factor-κB (NF-κB) is a highly conserved multifunctional transcription factor family, which has been widely studied due to its different roles in the development of tumours. NF-κB includes five members, namely REL (c-REL), RELA (p65), RELB, NFκB1 (p50) and NFκB2 (p52) (Ref. Reference Sun, Chang and Jin1). They form different homodimers or heterodimers involved in transcriptional regulation of many genes and play roles in immune, inflammation, oxidative stress, angiogenesis, tumour formation, cell proliferation and apoptosis (Refs Reference Bottex-Gauthier2, Reference Zhang, Lenardo and Baltimore3, Reference Taniguchi and Karin4). Angiogenesis is the formation of new blood vessels from the existing vascular system, which is an integral part of normal and pathological processes (Refs Reference Delgado5, Reference Geudens and Gerhardt6). Blood vessels are stationary or dormant in mature organisms, but still maintain important homeostasis. Only under certain physiological or pathological stimuli can they be reactivated. Angiogenesis is a key process that leads to neovascular diseases and required by tumour growth and metastasis. This is a complex and highly ordered process that relies on a broad signal transduction network between and within endothelial cells (ECs), related parietal cells (vascular smooth muscle cells and pericytes) and other cell types (such as immune cells) (Refs Reference Polyak, Haviv and Campbell7, Reference Polyak and Weinberg8, Reference Quail and Joyce9). In recent years, the molecular and cellular mechanisms involved in the regulation of angiogenesis and vascular homeostasis have been extensively studied. In addition to the common Notch signalling (Refs Reference Rehman and Wang10, Reference Fernández-Chacón11), studies in the past decades have revealed that NF-κB signalling pathway is involved in regulating angiogenesis and intravascular homeostasis under pathological conditions (Refs Reference Zhou12, Reference Noort13, Reference Ashander14). However, the mechanism and treatment strategy of NF-κB in human vascular diseases, especially cancer, are still controversial. This review covers the general theme of the biological role and mechanism of NF-κB in vascular-related diseases, and explores how to use NF-κB as a therapeutic target or mediator for diseases.
Structures of NF-κB and IκB proteins
NF-κB is a protein with multiple transcriptional regulation. NF-κB can be found in almost all mammalian cells, which regulates the production of a variety of encoding cytokines, growth factors, cell adhesion molecules and some acute protein factors, and plays an important role in inflammatory response, stress response, activation, proliferation, differentiation, apoptosis and tumour formation of immune cells (Refs Reference Bottex-Gauthier2, Reference Zhang, Lenardo and Baltimore3, Reference Taniguchi and Karin4). In mammals, NF-κB superfamily consists of five transcription factors: REL (c-REL), RELA (p65), RELB, NFκB1 (p50) and NFκB2 (p52) (Ref. Reference Sun, Chang and Jin1). Their N-terminal region has a highly conserved Rel homology domain (RHD). RHD is composed of N-terminal domain and C-terminal domain (CTD), and there is a nuclear-localisation sequence (NLS) on CTD, which is responsible for DNA binding, dimerisation and nuclear translocation. According to different structures, functions and synthesis methods, NF-κB superfamily is usually divided into two subfamilies: NF-κB subfamily (p50 and p52) and REL subfamily (c-REL, RELA and RELB). NF-κB subfamily is formed by the cleavage of precursor proteins p105 and p100 containing C-terminal ankyrin repeat domains (ARDs), respectively. These proteins contain RHD, but lack transcriptional activity region and unable to independently activate gene transcription unless they form heterodimeric complexes with REL subfamily proteins (Refs Reference Ghosh15, Reference Smale16). In addition to the N-terminal RHD, the C-terminal of REL subfamily contains transcriptional activation domain, rendering them capable of activating gene transcription directly. Except for RELB, other members of NF-κB can form all combinations of heterodimers and homodimers (Ref. Reference Neumann and Naumann17). However, RELB can only form heterodimers composed of p50 or p52. Whether an NF-κB dimer activates or inhibits transcription depends on the binding DNA region and the interaction with other transcription factors. NF-κB transcription factors can also play an indirect role in DNA regulatory elements when interacting with other DNA-binding transcription factors (Refs Reference Mulero18, Reference Ji19).
IκB is an important protein regulating the activity of NF-κB dimer, with 5–8 ARDs composed of 30 amino acids. The activation of NF-κB depends on the degradation of IκB protein. The IκB protein family has nine members including IκBα, IκBβ, IκBɛ, IκBζ, IκBη, Bcl-3, IκBNS, p100 and p105 (Ref. Reference Schuster20). The ARD at the N-terminal of IκB can bind to the RHD at the C-terminal of NF-κB dimer, covering the NLS of NF-κB to prevent NF-κB from moving into the nucleus (Refs Reference Sun21, Reference Karin and Greten22). The proline-glutamate-serine-threonine-rich motif at the C-terminal of IκB mainly maintains its structural stability and inhibits NF-κB binding to DNA (Refs Reference Majdalawieh and Ro23, Reference Ferreiro and Komives24). IκB kinase (IKK) complex phosphorylates the inhibitory protein IκBα, then IκBα is ubiquitinised and degrades by proteasome-dependent pathway. The released NF-κB is transcribed into the nucleus to activate the target gene (Refs Reference Karin and Greten22, Reference Chen and Greene25, Reference Israël26).
Activation of NF-κB
A large number of experiments have shown that a variety of factors could cause the activation of NF-κB signal transduction pathway. There are two widely accepted pathways: the canonical and non-canonical pathway, which are driven by the degradation of IκB or the induction of NF-κB-induced kinase (NIK), respectively (Fig. 1).

Figure 1. Activation of canonical and non-canonical NF-κB pathways. The canonical NF-κB pathway (left) is triggered by a variety of inflammatory factors and mediated by activation of the IKK complex, leading to transient nuclear translocation of NF-κB heterodimers. The non-canonical NF-κB pathway (right) is stimulated by specific receptors, dependent on NIK and IKKα, and mediates the sustained activation of the RelB/p52 complex.
IKK is composed of a large protein kinase complex with a size of 700–900 kDa, including three subunits, namely, two homologous catalytic subunits IKKα (IKK1) and IKKβ (IKK2) and an essential auxiliary subunit IKKγ (NEMO) with regulatory function (Ref. Reference Vallabhapurapu and Karin27). These two catalytic kinases, IKKα and IKKβ, are involved in different signalling pathways of NF-κB. The canonical NF-κB activation pathway is mainly regulated by IKKβ, while the regulation of the non-canonical NF-κB activation pathway is completely dependent on IKKα. Actually, the oligomers formed by IKKα/β/γ are found to affect the activity of IKKβ (Refs Reference Israël26, Reference Ruland28). IKKα can phosphorylate Ser32 and Ser36 on IκBα, while IKKβ not only phosphorylates Ser32 and Ser36 on IκBα, but also phosphorylates Ser19 and Ser23 on IκBβ. IKKγ with regulatory function has two extended α-helix regions and a zinc finger domain. Although IKKγ has no catalytic activity, the activity of IKK in canonical NF-κB pathway depends on the integrity of IKKγ subunits (Refs Reference Rothwarf and Karin29, Reference May, Marienfeld and Ghosh30, Reference Karin and Delhase31).
In the resting state of cells, p50/p65 or p50/c-REL in the cytoplasm binds to IκB to form a trimer, so that NF-κB dimer cannot be translocated. In the canonical NF-κB pathway, after the cells are stimulated by extracellular signals such as pro-inflammatory cytokines, lipopolysaccharide (LPS), growth factors and antigen receptors, IKKβ subunit is phosphorylated and activated, and then the Ser32 and Ser36 sites of IκBα are phosphorylated. The phosphorylated IκBα is further ubiquitinised and degraded in the 26S protein hydrolase complex. The released p50/p65 or p50/c-REL is transported into the nucleus to induce the expression of target genes (Ref. Reference Zhang32). The activation of canonical NF-κB pathway usually occurs in the early and transient process of immune activation (Ref. Reference Pires33).
Compared with the canonical NF-κB pathway, the non-canonical NF-κB pathway activators are relatively less, mainly including ligands for some members of the tumour necrosis factor (TNF) receptor family, such as lymphotoxinβ receptor (LTβR), nuclear factor receptor activator B receptor activator (RANK), CD40 and B-cell activating factor family (Refs Reference Cildir, Low and Tergaonkar34, Reference Maubach35, Reference Sun36, Reference Bonizzi and Karin37, Reference Vallabhapurapu38). In resting cells, TRAF2/3 recruits NIK into the complex, and subsequently cIAP1/2 ubiquitinates NIK through the ubiquitination of K48 connection, leading to the degradation of NIK protein. After extracellular signal stimulation, the TRAF2/TRAF3/cIAP1/cIAP2 complex is degraded, which prevents NIK protein degradation. The phosphorylation of IKKα complex is induced by NIK, thereby further activating p100, resulting in phosphorylation-dependent cleavage of p100. Subsequently, the active p52/RelB complex enters the nucleus and binds to the target gene to regulate gene expression (Ref. Reference Senftleben39). The activation of NIK is the key to initiate the downstream pathway, so the activation of non-canonical NF-κB pathway is slower but lasts longer. The non-canonical NF-κB pathway is mainly involved in long-term cellular processes, such as chronic inflammation (Refs Reference Vallabhapurapu and Karin27, Reference Sun36).
NF-κB-related microRNA in inflammation and angiogenesis
Due to the wide range of biological functions of this protein, the role of NF-κB in the development of human diseases has received increasing attention. Many miRNAs have been reported to be involved in inflammation and angiogenesis. MiRNAs are a class of non-coding single-stranded RNA molecules with a length of about 22 nucleotides, which play a regulatory role in target mRNA by damaging the stability of target mRNA and inhibiting the translation of target mRNA. Some of the miRNAs related to NF-κB have been implicated in inflammation and angiogenesis (Fig. 2). One group of miRNAs such as miR-17 and miR-31 in response to TNF-α inhibit intercellular adhesion molecule-1 (ICAM-1) expression and regulate angiogenesis by inhibiting leukocyte aggregation (Ref. Reference Doebele40). In addition, miR-31 was shown to promote the migration of ECs induced by Kaposi's sarcoma-associated herpes virus, while miR-17 had dual effects on angiogenesis in different tumours (Refs Reference Liu41, Reference Tsoyi42, Reference Sabatel43, Reference Zhu44). Another group of non-coding RNA upregulated by TNF-α, including miR-21, miR-221 and miR222, were proved to have anti-angiogenesis effect. In addition to inhibitory effect, miR-21 can also indirectly upregulate the expression of vascular cell adhesion molecule-1(VCAM-1) via targeting PTEN and promote inflammatory cell infiltration. In prostate cancer, miR-21 might be thought to promote angiogenesis (Refs Reference Ueda45, Reference Hu46). However, the increase of pre-miR-21 in the vitreous reduces the angiogenic response in the choroidal neovascularisation mouse model (Ref. Reference Suárez47). On the other hand, the expression of VCAM-1 stimulated by angiotensin II is partially downregulated by overexpressed miR-221 and miR-222, suggesting that these miR RNAs were involved in anti-inflammatory responses (Ref. Reference Poliseno48). They also target ICAM-1 and participate in the regulation of inflammatory cell infiltration (Refs Reference Zhu49, Reference Cheng50). Moreover, miR-221 and miR-222 were evidenced to inhibit EC proliferation and migration and downregulate endothelial nitric oxide synthases (eNOS) and c-kit expression (Refs Reference Sun51, Reference Fasanaro52). Similarly, miR-146, upregulated in ECs in response to NF-κB, represses endothelial activation by negatively regulating the expression of ICAM1, VCAM1 and E-selectin. However, there is opposite evidence that miR-146 also promotes angiogenesis by upregulating eNOS (Refs Reference Qi53, Reference Maijer54). Contrary to miR-21, miR-146, miR-221 and miR-222, the expression of miR-181b in ECs is downregulated by TNF-α treatment. MiR-181b inhibits NF-κB response genes such as VCAM-1 and E-selectin by targeting importin-α3 required for NF-κB nuclear translocation (Ref. Reference Madge55). Available data convinced that decreased oxygen partial pressure could induce miR-210 expression in ECs through HIF-1α-dependent pathway (Refs Reference Brown, Claudio and Siebenlist56, Reference Tak and Firestein57). MiR-210, known as master hypoxamiR, can be induced by HIF-1α to regulate a series of intracellular functions in response to hypoxic stress, thereby promoting cell survival and differentiation. On one hand, overexpression of MIR-210 promotes the migration of EC and increased the formation of capillary-like structure under hypoxia. On the other hand, miR-210 inhibits p50 protein and negatively regulated the expression of angiogenesis-promoting cytokines induced by NF-κB, such as IL-6 and TNF-α.

Figure 2. Multiple mediators are shared in the molecular pathways of pro-inflammatory response and angiogenesis. TNF-α induces NF-κB signalling pathway to initiate inflammatory response and HIF-1α transcription factor involved in angiogenesis is regulated by NF-κB under hypoxic condition. MicroRNAs regulate inflammation-related angiogenesis by acting on the downstream signalling molecules in NF-κB-mediated pro-inflammatory responses. In addition, the Nrf2/HO-1 axis that interacts with HIF-1α can regulate hypoxia-induced angiogenesis by targeting VEGF.
Apart from the microRNA in inflammation-related angiogenesis, the transcription factors in NF-κB are also closely related to inflammatory angiogenesis. Noort et al. reported that non-canonical NF-κB signal could regulate inflammation-induced angiogenesis (Refs Reference Noort13, Reference Suárez58). They demonstrated that NIK was highly expressed in inflammatory synovial ECs of rheumatoid arthritis (RA). In synovial ECs, NIK upregulates the expression of C-X-C motif chemokine 12 (CXCL12) through non-canonical NF-κB signalling pathway, which promoted pathological angiogenesis (Ref. Reference Tsai59). Thus, NIK is considered to be one of the key regulators of angiogenesis induced by synovitis in RA (Refs Reference Wu60, Reference Shan61).
Mechanisms of the constitutive activation of NF-κB in different cancers
A proper understanding of NF-κB regulatory mechanisms would certainly help to clarify its role in diseases, especially in tumoural progression. In most cancers, activation of NF-κB is enhanced and this structural activation occurs in a large number of different cancers through several mechanisms. Firstly, NF-κB activity can be activated by mutations in tumour-associated genes, a form generally associated with haematological malignancies (Refs Reference Vrábel, Pour and Ševčíková62, Reference Eluard63, Reference Lim, Yang and Staudt64). These mutations, which affect canonical or non-canonical NF-κB signalling, can occur in both core NF-κB pathway proteins and upstream regulatory proteins of NF-κB. On the one hand, the NF-κB transcription factor itself is the target of mutation, among them REL, NFκB1 and NFκB2 are more frequently mutated. Oncogenic mutations in NF-κB signalling proteins result in C-terminal truncations or single amino acid mutations, which enhance NF-κB activity (Ref. Reference Courtois and Gilmore65). Complete loss-of-function mutation of the IκB protein results in chronic nuclear localisation of NF-κB in glioblastoma, diffuse large B-cell lymphoma (DLBCL), Hodgkin's lymphoma and nasopharyngeal carcinoma (Refs Reference Lake66, Reference Kalaitzidis67, Reference Bredel68, Reference Zheng69). On the other hand, the most common mutations that enhance NF-κB signalling in DLBCL and multiple myeloma may occur in upstream molecules or downstream target genes of specific signalling pathways. Continued activation of canonical or non-canonical NF-κB driven by these molecular mutations is required for the proliferation and survival of malignant B cells (Refs Reference Vrábel, Pour and Ševčíková62, Reference Matthews70, Reference Young71). Secondly, cytokines or other stimulus-dependent activations of the NF-κB pathway can increase the constitutive activation of NF-κB via an autocrine mechanism. For example, TNF-α, encoded by NF-κB target gene TNFA, is a strong activator of NF-κB and acts in an autocrine manner on some tumour cells with constitutive activation of NF-κB (Ref. Reference Schmitz72). Thirdly, epigenetic status or epigenetic regulatory factors interact with the chronically active NF-κB found in many cancers, which makes the induced NF-κB preferentially bind to the genomic position that initiates epigenetics. NF-κB can also recruit chromatin modification coactivators such as histone acetyltransferase and histone deacetylase to further modify local chromatin (Refs Reference Mulero18, Reference Bhatt and Ghosh73, Reference Dawson and Kouzarides74). In addition, mutations in non-coding sequences cause carcinogenesis by affecting transcription and gene expression (Ref. Reference Patel and Wang75). For example, promoter sequence mutations affecting the expression of NF-κB and its target genes have been found in oral cancer, breast cancer and bladder cancer (Refs Reference Lin76, Reference Kang77). Finally, several oncogenic human viruses also affect NF-κB signal transduction. These viruses replicate and cause cancer by chronic NF-κB-dependent inflammation or the sustained activity NF-κB viral activators (Refs Reference Costa RM78, Reference Charostad79, Reference Harhaj and Giam80, Reference Shokri81).
The effect of NF-κB in different aspects of cancer progression
Tumour cell apoptosis progression affected by the activation of NF-κB
In tumour cells, activation of NF-κB usually results in inhibition of apoptosis. NF-κB target genes are known to encode anti-apoptotic molecules, such as BCL2, BCLXL and apoptosis suppressors. Increased expression of anti-apoptotic target genes is detected in a variety of tumour cells with constitutive NF-κB activity (Ref. Reference Gilmore82). Furthermore, the activated NF-κB increases the number of DNA damage cells that accumulate carcinogenic mutations by preventing P53-dependent apoptosis (Ref. Reference Gudkov, Gurova and Komarova83). In addition to anti-apoptosis, there are upregulated target genes related to cell proliferation in NF-κB signalling pathway, such as cyclin D1/D2/D3. Moreover, NF-κB also affects cell proliferation by regulating key enzymes. A variety of studies have shown that the activation of NF-κB in inflammatory immune cells stimulates the expression of inflammatory cytokines such as TNF-α and IL-6, which makes tumour cells and tumour stromal cells proliferate (Refs Reference Cerhan84, Reference Becker85). Additionally, it has been proved that NF-κB is a necessary downstream pathway for RAS oncogene activation that causes cell proliferation (Refs Reference Bassères86, Reference Finco87).
Cancer stem cells regulated by NF-κB in various cancers
NF-κB is involved in the functional regulation of cancer stem cells (CSCs). Studies have found that NF-κB is structurally activated in leukaemia, glioblastoma, breast cancer, ovarian cancer, colon cancer and other CSCs, which mediates tumour inflammation, cell proliferation, survival and maintenance (Ref. Reference Vazquez-Santillan88). In breast cancer, NF-κB activation mediated by Notch signalling pathway induces CSC proliferation, and the highly expressed NIK can induce the activation of the non-canonical NF-κB pathway to regulate CSC self-renewal and metastasis (Refs Reference Yamamoto89, Reference Vazquez-Santillan90). In ovarian cancer, CD44+ CSCs enhance the activity of p50/RelA dimer by upregulating core NF-κB signalling protein, thereby promoting self-renewal and metastasis of tumour cells (Ref. Reference Gonzalez-Torres91). In pancreatic cancer, the expression of transcription factor Sry-related HMG box 9 is enhanced by activating the typical NF-κB pathway, resulting in increased CSCs and enhanced invasiveness (Ref. Reference Sun92). In colorectal cancer, inflammatory mediator prostaglandin E2 promotes the formation, survival, proliferation and metastasis of colorectal CSCs by activating NF-κB pathway. The transcription factor Foxp3, however, inhibits the expression of the NF-κB target gene, which is associated with decreased colorectal CSCs and invasiveness (Refs Reference Liu93, Reference Wang94).
NF-κB and tumour microenvironment involved in cancer progression
The development and metastasis of tumours are inseparable from the tumour microenvironment composed of multiple stromal cells, tumour cells, cytokines, chemokines and so on. TNF-α is one of the most common tumour-promoting cytokines, mainly produced by activated neutrophils and macrophages, inducing the expression of other proinflammatory cytokines and promoting tumour progression (Refs Reference Balkwill95, Reference Grivennikov and Karin96). IL-6 is a cytokine dependent on NF-κB in tumour microenvironment, which is mainly produced by malignant tumour cells and activated immune cells. The activation of IL-6 is related to inflammatory tumour and stem cell amplification (Refs Reference Taniguchi and Karin97, Reference Chang, Daly and Bromberg98). Another important cytokine is transforming growth factor-β (TGF-β), which can induce naive T cells to differentiate into Treg and TH17 cells, and enhance tumour metastasis and invasion (Refs Reference Meulmeester and Ten Dijke99, Reference Grivennikov, Greten and Karin100). Besides cytokines, tumourigenesis is also associated with NF-κB-driven chemokine in the tumour microenvironment. The chemokines induced by NF-κB recruit more immune cells, thereby stimulating the inflammatory response, and play an important role in promoting tumour growth and metastasis, synthesising growth factors and inducing angiogenesis. For instance, in human and mouse pancreatic tumour models, CCR5/CCL5 signal interference reduces the migration of Treg cells to tumour microenvironment, indicating that chemokine chemokine (C-C motif) ligand 5 (CCL5) is necessary for Treg cell migration (Ref. Reference Tan101). CCL5 induces the formation of eukaryotic initiation factor 4F translation initiation complex and upregulates the expression of cyclin D1, c-Myc and anti-cell death defence protein-1, thereby promoting the proliferation of breast cancer cells (Ref. Reference Murooka, Rahbar and Fish102). Meanwhile, macrophages are widely enriched in tumour microenvironment and activate cytokines by producing inflammatory factors, chemokines and proteases such as cysteine cathepsins, thereby promoting tumour progression (Refs Reference Quail and Joyce9, Reference Grivennikov, Greten and Karin100). Since M1 macrophages polarized to M2 macrophages through IL-1R and MyD88 requires the participation of the canonical NF-κB signalling pathway, inhibition of the NF-κB can increase the production of M1 macrophages with anti-tumour cytotoxicity (Ref. Reference Hagemann103). Dendritic cell (DC) is known to be the most powerful antigen-presenting cell and plays an important role in anti-tumour immunity. The experiment showed that PD-1 inhibited the anti-tumour function of DCs by inactivating NF-κB in infiltrating DCs of ovarian tumours (Refs Reference Karyampudi104, Reference Antonangeli105). In natural killer cells with antitumor activity, the expression of cytotoxicity effector is also affected by NF-κB (Ref. Reference Vivier106). In tumour immunity, Treg cells can secrete a variety of inhibitory cytokines, limiting the ability of the immune system to kill tumours and allowing tumour cells to escape immune surveillance. The maturation and maintenance of immune tolerance of Tregs require the involvement of p65 and Rel (Refs Reference Oh107, Reference Isomura108, Reference Evaristo109). It ss well known that T-cell receptor and B-cell receptor are one of the important stimuli that induce the activation of NF-κB. T and B cells regulated by NF-κB pathway play a pro-tumour or anti-tumour role in the tumour microenvironment (Ref. Reference Ward, Gubin and Schreiber110). Cancer-associated fibroblasts (CAFs) are one of the most important components in the tumour microenvironment and play an essential role in the development of tumours. They have multiple functions, including extracellular matrix (ECM) remodelling, metabolism, angiogenesis regulation and interactions with cancer cells and invasive immune cells through the production of growth factors, cytokines and chemokines (Refs Reference Kalluri111, Reference Koliaraki112). In the early stage of tumourigenesis, tumour-derived IL-1β induces CAF activation and promotes tumour inflammation in a NF-κB-dependent manner (Ref. Reference Erez113). Through the study of pancreatic cancer, it was found that CAF promoted the production of cytokines such as GM-κ and IL-6 with the participation of NF-κB, which induced extracellular metastasis and paracrine of tumour cells (Ref. Reference Santolla114).
The relationship between inflammation and tumour has been confirmed in cancer research. Chronic inflammation leads to the occurrence and development of malignant tumours by producing a large number of cytokines and recruiting a variety of immunosuppressive cells (Ref. Reference Taniguchi and Karin4). The activation of NF-κB signalling pathway is crucial in regulating inflammatory response. Early studies have shown that NF-κB plays a key role in inflammation-driven colitis-related cancer and hepatocellular carcinoma (Refs Reference Greten115, Reference Pikarsky116). In the mouse model of colitis-related cancer, the loss of IKKβ inhibits the occurrence of colon inflammation and colitis-related cancer. Some of the viruses, such as hepatitis B virus, Kaposi sarcoma herpesvirus and human papillomavirus, increase cell stress response and recruit inflammatory immune cells through chronic NF-κB-dependent inflammatory reactions, thereby leading to malignant tumours. There are also some viruses that directly encode proteins that activate the NF-κB signalling pathway (Refs Reference Costa RM78, Reference Charostad79, Reference Karin117). For example, in B-cell lymphoma and nasopharyngeal carcinoma, EB virus through the activation of NF-κB encodes the protein of LMP1 as a CD40 homologue to prevent the apoptosis of B cells infected by EB virus (Ref. Reference Charostad79). Another mechanism by which NF-κB stimulates tumourigenesis is to induce DNA damage. Chronic inflammation and NF-κB can not only cause DNA damage and carcinogenic mutations by promoting the production of reactive oxygen species and reactive nitrogen, but also lead to chromosomal instability, aneuploidy and epigenetic changes (Refs Reference DiDonato, Mercurio and Karin118, Reference Joyce119, Reference Kiraly120).
The potential tumour suppressor function of NF-κB
Despite the overwhelming evidence that NF-κB is oncogenic, studies have revealed its potential for tumour suppressor function. Since NF-κB can induce the expression of a variety of proapoptotic molecules, such as Bcl-xs, Bax, Fas or FasL, NF-κB is considered to promote apoptosis and may be a tumour suppressor gene (Ref. Reference Dutta121). In the epithelial tissue, downregulated NF-κB by overexpression of IκBα leads to skin epidermis hyperplasia in mice, indicating that NF-κB activation is important for cell growth inhibition. RelA-deficient mouse cell lines have a weak transformation phenotype, and RelA-deficient fibroblasts in immunodeficient mice have the ability to form tumours, indicating that RelA has tumour inhibitory activity in some cases, and RelA complex is involved in the control of various cell characteristics related to tumourigenesis (Ref. Reference Gapuzan, Yufit and Gilmore122). The anti-tumour function of NF-κB can also be achieved by participating in the activation of conventional T cells via the canonical pathway, which is necessary for CD8+T cell proliferation and anti-tumour immune response (Ref. Reference Gerondakis123). Studies have shown that LTβR-mediated non-canonical NF-κB activation induces the expression of adhesion molecules and chemokines, and ultimately forms tertiary lymphoid tissues. It is worth noting that the presence of tertiary lymphoid tissues in the tumour microenvironment generally suggests a better prognosis (Refs Reference Lin124, Reference Furtado125, Reference Luther126, Reference Sautès-Fridman127). Moreover, there is a complicate cross-signalling between NF-κB and wild-type/mutant tumour suppressor gene p53, which indicates that the NF-κB pathway is possibly involved in the mechanism of tumour suppression (Ref. Reference Carrà128). Although the above studies have shown that NF-κB may inhibit tumours by promoting apoptosis, enhancing immune function and regulating tumour microenvironment, there is still no report clearly indicating the inhibitory effect of NF-κB on different cancers in clinical research. Based on the current research, the activation of NF-κB has the potential to promote the occurrence and development of haematological tumours and most solid tumours including glioblastoma, nasopharyngeal carcinoma, breast cancer, ovarian cancer, colon cancer and pancreatic cancer.
NF-κB signalling pathway promotes pathological angiogenesis by activating endothelial cells and stimulating basement membrane degradation
Combined with the above reports, NF-κB signalling pathway plays an important role in the regulation of pathological angiogenesis, especially tumour angiogenesis (Fig. 3). Kei et al. found that in colorectal cancer, the abnormal activation of NF-κB signal was positively correlated with the increased number of pathological angiogenesis (Ref. Reference Sakamoto129). In fact, the positive effect of NF-κB on tumour angiogenesis is closely related to inflammatory response. NF-κB activated by inflammatory cytokines such as TNF, IL-1 and IL-6 can induce the expression of various pro-inflammatory genes and adhesion molecules in ECs, which in turn promoted the accumulation of inflammatory cells in perivascular matrix and thus enhanced the activation of NF-κB (Ref. Reference Ashander14). Meanwhile, in ECs activated by inflammatory factors, NF-κB can be phosphorylated and nuclear translocated, which promotes the transcription of many angiogenesis genes. Vascular endothelial growth factor (VEGF) is considered as a key medium for angiogenesis in tumours. Under the regulation of activated NF-κB, ECs, macrophages and tumour cells were shown to secrete VEGF, which was also one of the NF-κB-regulated target genes (Refs Reference Lennikov130, Reference Shen131). VEGFR2 is the main receptor of VEGF and the signal pathway activated by VEGF/VEGFR2 interaction plays a leading role in angiogenesis. It was found that NF-kB binds directly to the upstream promoter of VEGFR2 and activated its transcription. Once activated VEGFR2 binds to VEGF, VEGFR2 would activate a series of downstream mediators to promote angiogenesis (Ref. Reference Dong132).

Figure 3. The positive regulation mechanism of NF-κB on angiogenesis by transcription target gene. Under the action of inflammatory cytokines, NF-κB induces the expression of various pro-inflammatory genes and adhesion molecules in endothelial cells, which in turn promoted the accumulation of inflammatory cells in perivascular matrix and enhanced the activation of NF-κB. Activated NF-κB can not only make endothelial cells directly secrete VEGF, but also bind to the upstream promoter of VEGFR2 and activate its transcription. Once VEGFR2 binds to vascular endothelial growth factor, VEGFR2 activates a series of downstream mediators to promote angiogenesis. In addition, activated NF-κB can stimulate the expression of matrix metalloproteinases, promote the degradation of basement membrane and extracellular matrix. Nuclear transcription of NF-κB also upregulates inflammatory chemokines IL-6 and IL-8, thereby stimulating the proliferation and migration of tumour endothelial cells. Meanwhile, activated NF-κB can promote tumour angiogenesis by upregulating MCP-1 and COX-2. In non-canonical NF-κB pathway, NIK upregulates the expression of chemokine CXCL12, which plays an important role in the homing of endothelial progenitor cells.
It is well known that one of the key steps of tumour angiogenesis is the degradation of vascular basement membrane and ECM. The activated NF-κB can stimulate the expression of multiple matrix metalloproteinases, including MMP2, MMP3 and MMP9, thereby promoting the degradation of basement membrane and ECM (Refs Reference Mountain133, Reference Popov134, Reference Ko135). In addition, the study also found that the nuclear transcription of NF-κB upregulates inflammatory chemokine IL-8, which can activate ECs of tumour vessels and induce tumour-associated macrophages to secrete additional growth factors to promote tumour invasion and migration. Interestingly, IL-8 also induces NF-κB activation by binding to its homologous receptor C-X-C motif chemokine receptor 2 (CXCR2), which upregulates VEGF gene and protein levels in ECs, leading to autocrine activation of VEGFR2 (Refs Reference Martin, Galisteo and Gutkind136, Reference Passaro137). In addition, the activation of NF-κB can also upregulate the expression levels of angiogenesis factors, including MCP-1 and COX-2. MCP-1 stimulates tumour infiltrating immune cells to secrete multiple angiogenesis-related factors by recruiting macrophages into tumour microenvironment (Ref. Reference Chen138). COX-2 promotes tumour growth by upregulating the production of multiple angiogenesis-related proteins (Ref. Reference Chen and Liu139). Wang et al. found that in colorectal cancer, B7-H3 could upregulate the expression of VEGF-A and promote angiogenesis by activating the NF-κB pathway. However, NF-κB interacting long non-coding RNA inhibits EC proliferation, migration and angiogenesis by inhibiting the secretion of IL-6 via NF-κB signalling pathway in breast cancer (Ref. Reference Luo140). In prostate cancer, NF-κB inactivation significantly inhibits the expression of proangiogenic molecules, including VEGF, IL-8 and MMP-9 in vitro and in vivo, thereby reducing tumour angiogenesis (Ref. Reference Huang141). NF-κB has also been implicated in epithelial-mesenchymal transition (EMT) in tumour cells. NF-κB can regulate EMT transcription factors, mainly including Twist, zinc finger transcription factor Snail2 (Slug) and Smad interaction protein 1, which initiate EMT and exert the ability of self-renewal by enhancing the stemness and invasiveness of cancer cells (Refs Reference Pires142, Reference Scheel and Weinberg143). In addition to EMT transcription factors, inhibition of NF-κB activity leads to decreased activity of various substrate degradation enzymes that promote EMT. In the progression model of breast cancer, IKK-β/IκBα/NF-κB pathway is necessary to induce and maintain EMT, so the activation of NF-κB is crucial to the metastasis potential of epithelial cells in breast cancer (Ref. Reference Huber144). Since MMPs induced by NF-κB stimulate the production of TGF-β, which promotes EMT process and tissue hypoxia, and ultimately lead to tumour cell metastasis and proliferation, TGF-β-induced EMT is at least partially dependent on NF-κB (Refs Reference Malki145, Reference Balkwill146). Additionally, NF-κB combined with some cytokines can directly target metastasis-related genes to control tumour cell motility.
It is noteworthy that under hypoxic environment, hypoxia-inducible factor-1α (HIF-1α) pathway plays an important auxiliary role in the regulation of inflammation-related angiogenesis genes (Refs Reference Cummins147, Reference Oliver, Taylor and Cummins148). In standard conditions, prolyl hydroxylases (PHDs), the main protease that degrades HIF in vivo, hydroxylates IKKβ to make it inactive, resulting in the inactivation of NF-κB in cytoplasm. In hypoxic tumour microenvironment, however, hypoxia causes PHDs to fail, and IKKβ activity cannot be inhibited, which leads to NF-κB activation and released to the nucleus. Since the NF-κB binding site is located in the promoter region of HIF-1α, activated NF-κB can directly regulate the expression of HIF-1α (Fig. 4) (Refs Reference Bonello149, Reference Frede150, Reference Jung151). In the ECs of renal ischaemia/reperfusion model, NF-κB induced by LPS regulates the expression of HIF-2α, which increases the production of NO, thereby improving renal microvascular perfusion and reducing ischaemic renal tubular injury (Fig. 4) (Ref. Reference He152). Another important mechanism involved in the hypoxia-related angiogenesis is NF-E2-related factor 2 (Nrf2)/haeme oxygenase-1 (HO-1) pathway. As a key transcription factor involved in cell response to oxidative and electrophilic stress, Nrf2 plays an important role in inducing antioxidant response (Ref. Reference Bellezza153). HO-1 is one of the most important downstream regulatory products of Nrf2, which can catalyse haeme to produce biliverdin, carbon monoxide and iron. Biliverdin is converted into bilirubin by biliverdin reductase, both of which are powerful free radical scavengers in the body (Ref. Reference Maines154). Under hypoxic conditions, HIF-1α and Nrf2 stress response pathways exist in a complex, interacting signalling network. HIF-1α is a transcriptional activator of oxygen-sensitive genes and is involved in hypoxia-induced HO-1 expression. In recent years, the cascade reaction of Nrf2 and HO-1 is considered to be one of the most important intracellular antioxidant stress mechanisms (Ref. Reference Rubiolo, Mithieux and Vega155). HO-1 has been found to mediate the synthesis and activity of the important pro-angiogenic factor VEGF, forming a positive feedback mechanism (Ref. Reference Józkowicz156) (Fig. 2).

Figure 4. NF-κB promotes angiogenesis under different pathological conditions. In the hypoxic tumour microenvironment, hypoxia renders PHDs that inhibit IKKβ activity ineffective. Activated NF-κB is transported to the nucleus and induces HIF-1α transcription, which promotes the expression of angiogenic factors. In endothelial cells of renal ischaemia/reperfusion model, LPS-induced NF-κB regulates HIF-2α expression and increases NO production, thereby improving renal microvascular perfusion.
In addition to canonical NF-κB signalling pathway, non-canonical NF-κB pathway is also involved in tumour progression (Fig. 3), but its effect on angiogenesis is still not comprehensive. Unlike canonical pathway, non-canonical pathway does not require the participation of IKKβ, but NIK and IKKα are indispensable. Previous studies have shown that NIK-mediated non-canonical NF-κB signalling pathway regulated inflammation-induced and tumour-related angiogenesis. NIK has been confirmed to be expressed in human melanoma, renal cell carcinoma, breast cancer, colorectal cancer and pancreatic cancer, and with the deletion of NIK gene, tumour-related angiogenesis was reduced (Ref. Reference Noort13). This effect is due to NIK involved in non-canonical NF-κB pathway can upregulate CXCL12, which is a chemokine that promotes angiogenesis and plays an important role in CXCR4 homing of endothelial progenitor cells. Of note, gene deletion of NIK only inhibits pathological angiogenesis, and does not affect the angiogenesis induced by TNF-α or VEGF during normal development.
NF-κB signalling pathway inhibits pathological angiogenesis by negatively regulating ECM degradation and endothelial cell activity
Paradoxically, the activation of NF-κB signalling pathway also inhibits angiogenesis (Fig. 5). In 2006, Kisseleva et al. reported that the transplanted tumours grew faster and the tumour vessels increased significantly in Tie2 promoter-IκBα transgenic mice, leading to the inhibition of NF-κB activity in ECs (Ref. Reference Kisseleva157). This study first demonstrated the inhibitory effect of NF-κB on tumour angiogenesis. Actually, it is not clear what mechanism NF-κB inhibits tumour angiogenesis and how NF-κB is activated in ECs. In general, angiogenesis requires a series of coordinated steps, in which the degradation of vascular endothelial matrix is a necessary process. NF-κB upregulates tissue inhibitors of metalloproteinase-1 (TIMP-1), a matrix metalloproteinases inhibitor that can prevent EC migration, in astrocytes by secreting IL-1. Similarly, in ECs, reactive oxygen species and TNF-α induce the expression of plasminogen activator inhibitor-1 (PAI-1) through NF-κB, and inhibit the activation of plasminogen to hydrolyse endothelial matrix (Ref. Reference Ricke-Hoch158). TNF-α can also inhibit the expression of tissue plasminogen activator (tPA) through NF-κB, which leads to the decrease of ECM degradation ability and inhibition of angiogenesis (Ref. Reference Ulfhammer159). In addition to basement membrane degradation, ECM remodelling is also a necessary condition for angiogenesis. Integrin is a cell adhesion and signalling protein that mediates the interaction between cells and ECM. The interaction between αVβ3-integrin and ECM has been shown to activate canonical NF-κB signal pathway (Refs Reference Rice160, Reference Jiang161, Reference Liu162). However, the role of the activated signal in ECM remodelling needs to be further explored. The binding site of NF-κB is located in the promoter regions of thrombin-sensitive proteins-1 and 2 (TSP-1 and 2), which were the earliest discovered natural vascular inhibitors (Ref. Reference Yang163). Later, vascular endothelial growth inhibitor (VEGI), which inhibits vascular EC proliferation and angiogenesis, has also been found to be induced by NF-κB (Ref. Reference Xiao164).
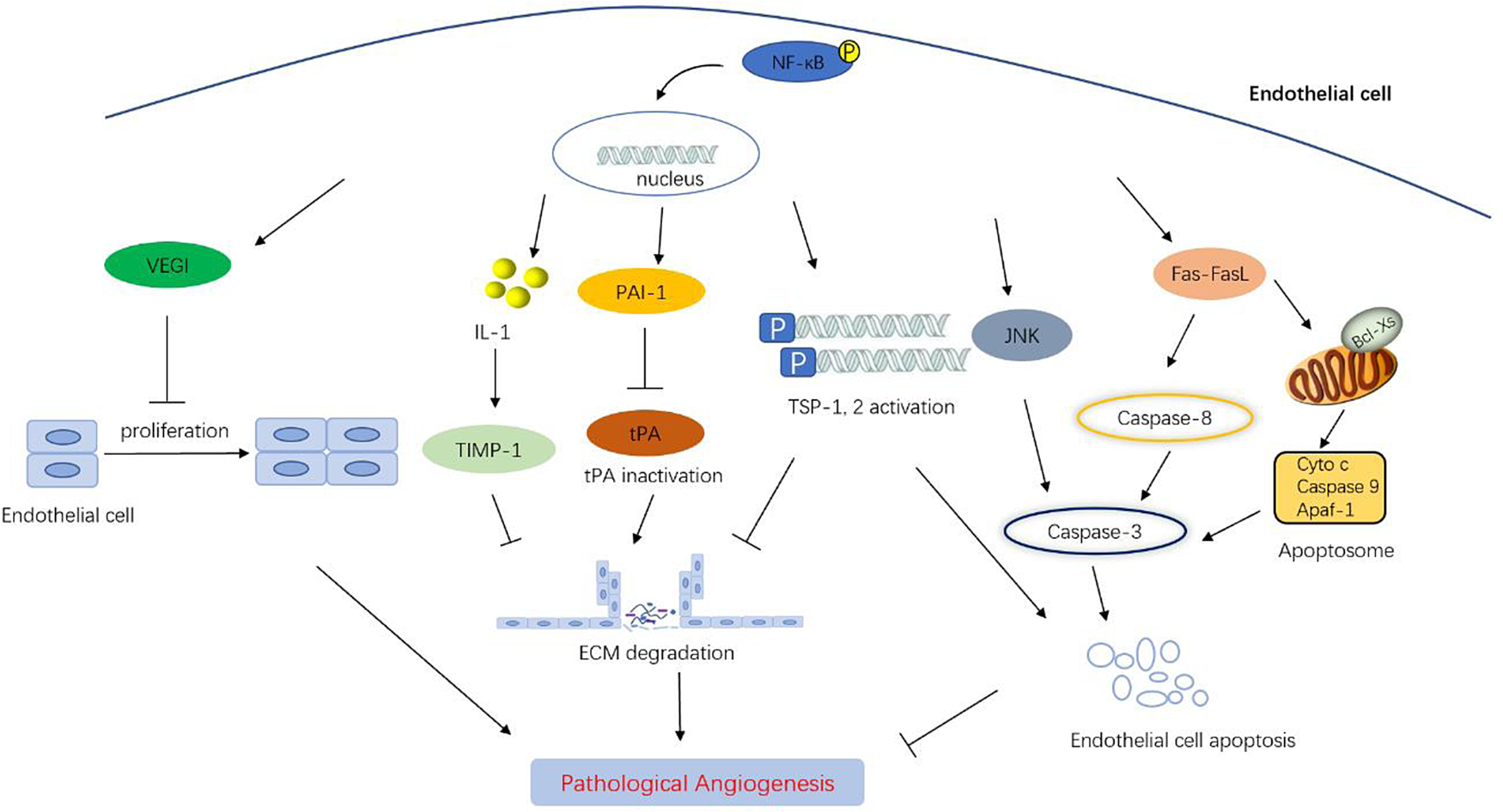
Figure 5. The negative regulation mechanism of NF-κB on angiogenesis by transcription target gene. On the one hand, NF-κB activates the expression of TIMP-1, PAI-1 and TSP-1, 2, resulting in decreased extracellular matrix degradation and hindering angiogenesis. On the other hand, NF-κB nuclear transcription can activate apoptosis signalling pathway, induce the expression of downstream pro-apoptotic genes such as JNK, Fas, FasL, caspase-3, thus promoting endothelial cell apoptosis. In addition, activated NF-κB induces the upregulation of VEGI, thereby inhibiting endothelial cell proliferation and growth.
A large number of evidences show that the activation of NF-κB is able to promote EC apoptosis. Under high glucose stimulation, PI3K/Akt/eNOS/NO pathway in HUVECs is activated to protect ECs from apoptosis at early stage. However, long-term stimulation with high concentration of glucose causes ECs to produce continuous and excessive reactive oxygen species, resulting in the continuous activation of NF-κB, and the downstream c-jun amino-terminal kinase (JNK) and caspase3 are also sequentially activated, which promotes EC apoptosis (Refs Reference Ho165, Reference Zhazykbayeva166). Furthermore, NO-mediated inhibition of NF-κB signal was reported to reverse cell apoptosis. IL-18 can induce the expression of proapoptotic genes by activating the NF-κB signalling pathway, including Bcl-Xs, Fas and FasL, leading to the apoptosis of endogenous and exogenous human cardiac microvascular endothelial cells (Ref. Reference Chandrasekar167). In addition, angiopoietin-1 was revealed to activate A20 binding inhibitor of NF-κB activation-2, an endogenous inhibitor of NF-κB, to protect ECs from apoptosis (Refs Reference Tadros168, Reference Chng169). Indeed, negative regulators that inhibit NF-κB activation were also shown to significantly reduce tube formation in HUVECs. Sprouty1 (SPRY1) is an inhibitor of MAPK pathway, and the regulation of SPRY1 expression is dependent of NF-κB signalling pathway. Some scholars have shown that SPRY1 promoted EC apoptosis, reduced EC proliferation, migration and adhesion as well as inhibited endogenous angiogenesis (Ref. Reference Sabatel170). Although silencing NF-κB can reverse the anti-apoptosis effect of apoptotic exosome-like vesicles (ApoExo) on ECs, inhibition of NF-κB restores the reduction of tube formation and CD31 expression in ECs induced by ApoExo (Ref. Reference Migneault171). Tumour angiogenesis is associated with uncontrolled gene expression of vascular ECs. Bromide domain containing 7 (BRD7) is considered to be a tumour suppressor gene, and the expression of BRD7 is negatively correlated with the activation of ECs (Refs Reference Park172, Reference Peng173). In ECs, the overexpression of BRD7 enhances NF-κB activity stimulated by bromine-dependent domain and upregulates the expression of NF-κB-dependent adhesion molecules (Ref. Reference van Beijnum174). However, the complex mechanism of BRD7 in NF-κB-mediated process has not been fully clarified. In addition to inhibiting tumour angiogenesis, the activation of NF-κB may also play an anti-tumour role through reversing endothelial anergy to inflammatory signals. Compared with normal ECs, tumour ECs are exposed to angiogenic growth factors and show reduced expression of adhesion molecules such as ICAM-1, ICAM-2 and VCAM-1. NF-κB has been found to upregulate multiple adhesion molecules in tumour ECs and restore leukocyte–endothelial interaction, resulting in anti-tumour immune effects. However, angiogenesis factors inhibit NF-κB pathway and the transcription of related adhesion molecules (Refs Reference Tromp175, Reference Luo176, Reference Dirkx177, Reference Yu178). These results suggest that NF-κB activated by vascular inhibitors not only directly negatively regulates ECs, but also indirectly exerts anti-tumour effect by upregulating the expression of adhesion molecules and reversing the inhibited inflammatory response.
NF-κB: a target for angiostatic therapy or angiogenic therapy?
It is well known that early activation of angiogenesis is a necessary condition for maintaining tumour cell proliferation. In addition to serving as a provider of nutrition, oxygen and waste transport, blood vessels also permit distant metastasis of tumour cells. The highly invasive process in tumours initiates with vasodilation and vascular permeability, causing extravasation of proteins that destroys the endothelial basement membrane (Ref. Reference Ghosh15). After the endothelial basement membrane of the vascular wall is destroyed, ECs, pericytes and vascular smooth muscle cells migrate to the direction of angiogenesis stimulation produced by tumour cells or host cells (Ref. Reference Armulik, Genové and Betsholtz179). The activated proliferating ECs adhere to each other to form tubular structures, accompanied by the formation of basement membrane and pericyte attachment. With the further maturation of the vascular network, capillaries are transformed into larger blood vessels, arteries and veins so that a new circulatory system is established (Ref. Reference Zuazo-Gaztelu and Casanovas180). In the tumour ecosystem, tumour cells and their microenvironment stimulate angiogenesis by regulating the expression of pro-angiogenic factors and/or anti-angiogenic factors to promote further tumour growth (Refs Reference Fraisl181, Reference Goel182).
Considering the important role of activated NF-κB in tumour angiogenesis, a lot of efforts have been made to explore strategies for NF-κB as a target to assist cancer prevention and treatment. Some NF-κB compound inhibitors that block the formation of new blood vessels in tumours have been reported. Since the cytokine IL-6 produced by NF-κB plays an important role in the formation of new blood vessels in tumour, NF-κB/IL-6 signal is considered as a promising target in tumour treatment. In triple-negative breast cancer (TNBC), the specific agonist of G protein-coupled oestrogen receptor activates ERK1/2 and PI3K/Akt in TNBC cells, and suppresses the phosphorylation of GSK-3β, leading to inhibit the transcription activity of NF-κB. The inactivated NF-κB reduces its binding to the IL-6 promoter, which allows tumour cell migration and angiogenesis to be hindered (Ref. Reference Chen183). STAT3 and HIF-1α are considered to be the main signal transduction molecules downstream of IL-6 signalling, and inactivation of HIF-1α and STAT3 can inhibit the occurrence and development of tumours. Liang et al. confirmed that inhibition of NF-kB/IL-6 was a prerequisite for inactivation of STAT3 and HIF-1α pathways induced by G protein-coupled oestrogen receptor-specific agonists. It is noteworthy that the activation of NF-κB/IL-6 seems also to play a positive role in cerebrovascular formation. Unlike anti-angiogenesis therapy in tumours, Zhuang et al. found that autophagy-related protein ATG7 homologous to ubiquitin-activated enzyme E1 mediated the production of IL-6 in cerebral microvascular ECs in a NF-κB-dependent manner and promoted angiogenesis in cerebral vessels (Ref. Reference Zhuang184).
Generally, many targets of NF-κB inhibitors ranging from cell surface receptors to nuclear signaling have been identified (Fig. 6) (Ref. Reference Ramadass, Vaiyapuri and Tergaonkar185). Among them, lots of inhibitors are extensively studied and undergoing clinic trials for treatment of cancer. Their effects on angiogenesis are partially examined. For example, the IKKa/b inhibitors including CHS-828, IMD-1041 and EB-1627 have been shown to reduce angiogenesis by downregulating CCL2, CXCL5, Cxcr2 and TNF-α (Refs Reference Lennikov130, Reference Huang186). BTK inhibitors such as ibrutinib, zanubrutinib and acalabrutinib are promising drugs for cancer treatment. A recent study shows that ibrutinib inhibits angiogenesis. Interestingly, the effect of ibrutinib on angiogenesis is independent on BTK but via stimulating more BMP4 expression (Ref. Reference Liu187). Another group of NF-κB inhibitors are inhibitors of proteasome, such as bortezomib and disulfiram. Both bortezomib and disulfiram have been shown to inhibit angiogenesis via reducing the production of VEGF, HGF and bFGF or weakening EGFR phosphorylation (Refs Reference Wang, Zhang and Yao188, Reference Li189). Selinexor is a potent and selective exportin-1 inhibitor that affects nuclear translocation of NF-κB. It also inhibits angiogenesis in tumour mouse models (Ref. Reference Gravina190). In nucleus, the activity of NF-κB is modulated by deacetylase and a group of deacetylase inhibitors including azacitidine, vorinostat, romidepsin, belinostat, panobinostat and decitabine can inhibit angiogenesis via downregulating VEGF and nitric oxide synthase gene (Ref. Reference Singh, Bishayee and Pandey191). Thus, small molecules among NF-κB inhibitors generally show inhibitory effect on angiogenesis.

Figure 6. Many NF-κB inhibitors show inhibitory effect on angiogenesis. The list of NFkB inhibitors of which the effects on angiogenesis have been examined in vitro and in vivo. The inhibitors including IKK inhibitors (CHS-828, IMD-1041 and EB-1627), BTK inhibitors (zanubrutinib, acalabrutinib and ibrutinib), proteasome inhibitors (disulfiram and bortezomib), nuclear translocation inhibitor (selinexor), histone deacetylase inhibitors (vorinostat, romidepsin, belinostat, panobinostat, azacitidine and decitabine) and natural products (curcumin, quercetin and neovastat).
Besides small molecules, there are also natural products that inhibit NF-κB (Fig. 6). As the most common natural NF-kB inhibitor, curcumin primarily induces tumour cell apoptosis and inhibits angiogenesis. The inhibitory effect of curcumin on angiogenesis is mainly through NF-κB-mediated downregulation of COX-2 and VEGF and other angiogenesis-related genes. This effect is reflected in the colon cancer xenograft model that curcumin significantly reduces tumour growth by inhibiting the expression of COX-2 and VGGF (Ref. Reference Kunnumakkara192). One study indicated that the inhibitory effect of curcumin on VEGF could inhibit the binding ability of NF-κB to DNA in human tongue squamous tumour cells transfected with luciferase gene, and enhance its sensitisation effect on treatment (Ref. Reference Chiang193). In addition, curcumin can increase the expression of apoptosis mediators such as cytochrome C, caspase-3 and caspase-8 in tumour cells. Curcumin inhibits p65 mediated by ERK1/2 and SAPK/JNK to limit the growth of androgen-independent prostate cancer cells, and curcumin also has the ability to effectively restrain stemness features of liver cancer cells (Refs Reference Li194, Reference Marquardt195). The substantial clinical trials on curcumin are underway or have been completed. A large number of clinical trials support the potential of curcumin for the treatment of various cancers (Ref. Reference Li and Zhang196). Considering that the bioavailability of curcumin is still generally low, the research and development of targeted formulations based on various carrier types has attracted increasing attention (Refs Reference Dhillon197, Reference Anand198).
Quercetin, a kind of flavonoids with chemical therapy and chemical prevention, can be found in some plants such as apples, grapes, onions, nuts and kale. This plant flavanol plays an important role in inhibiting tumour progression by regulating multiple signalling pathways. In serval tumours, quercetin can not only negatively regulate ECs and inhibit tumour angiogenesis, but also induce tumour cell apoptosis by inhibiting NF-κB and its downstream genes (Refs Reference Long199, Reference Lei200, Reference Huang201, Reference Granado-Serrano202). Several clinical trials have confirmed the safety of quercetin administration and observed its antitumor activity. Clinical experiments have shown that quercetin can inhibit the growth of prostate cancer tumours and reduce the risk of prostate cancer (Refs Reference McCann203, Reference Nieman204). Another clinical study evaluated the synergistic effect of quercetin and sorafenib in hepatocellular carcinoma (Ref. Reference Brito205). It is known that ovarian cancer stem cells (OCSCs) are transformed into vascular precursor cells after activation by NF-κB signalling pathway, which is the unique ability of OCSC. IKKβ inhibitor BAY11-7082 was reported to prevent ovarian cancer angiogenesis; however, this inhibitor had no effect on vascular development of normal ECs. These results showed that canonical NF-κB inactivation could inhibit OCSC differentiation and the ability to produce ECs (Refs Reference Vazquez-Santillan88, Reference Alvero206). Furthermore, kallistatin is a serine protease inhibitor that inhibits TNF-α-induced degradation of IκBα, phosphorylation of IKK and p65 protein and nuclear translocation of p65/50. Studies have shown that kallistatin may reduce the expression of some angiogenesis-related genes including VEGF via inhibition of the NF-kB signalling pathway, leading to anti-tumour angiogenesis (Ref. Reference Huang207). However, the potential application of kallistatin in tumours needs to be further verified in preclinical and clinical trials.
Paradoxically, the proliferation, migration and survival of ECs are found to be inhibited under the regulation of NF-κB, therefore the activation of NF-κB should have a dual effect on tumour angiogenesis. Several angiostatic compounds have been reported to activate NF-κB in ECs to prevent tumour growth. As an effective vascular inhibitor, the 16 kDa N-terminal fragment of prolactin (16 K PRL) inhibits the proliferation and migration of ECs. The16 K human PRL induces the activation of caspase-8, caspase-9 and subsequent cell apoptosis, which requires the activation of NF-kB (Refs Reference Tabruyn208, Reference Tabruyn209). In addition to the 16 kDa N-terminal fragment of prolactin, platelet factor 4 (PF4), a platelet-specific chemokine, inhibits EC proliferation, migration and angiogenesis in vitro and in vivo. Experiments and data showed that PF4 could increase the expression of E-selectin in ECs, and the cis-acting element sensitive to PF4 in E-selectin promoter was the binding site of NF-κB (Ref. Reference Yu178). These results suggested that PF4 induced E-selectin expression in ECs by activating NF-κB transcriptional activity. In order to analyse the changes of NF-κB-related gene expression after angiostatin treatment, researchers apply gene microarray analysis technology. Although there is no direct evidence that NF-κB is activated, angiostatin is associated with the increased expression of RELB and many NF-κB target genes, including E-selectin, cyclin D1, p21 and ICAM-1 (Ref. Reference Chen210). Furthermore, the use of statins has been reported to inhibit tumour growth and angiogenesis. Atorvastatin increases the production of reactive oxygen species through the activation of NF-κB, which leads to the loss of NO bioavailability, followed by the formation of strong oxidant peroxynitrite. The activation of NF-κB upregulates the expression of neuronal and inducible nitric oxide synthase capable of inhibiting angiogenesis and leading to vascular injury (Refs Reference Hindler211, Reference Nakata212). In addition, Gingras et al. demonstrated that neovastat, another angiogenesis inhibitor, suppresses angiogenesis by increasing tPA activity in a NF-κB-dependent manner (Refs Reference Gingras213, Reference Simard214). Clinical trials of neovastat in multiple myeloma, plasmacytoma, breast cancer, lung cancer, kidney cancer or colon cancer have been completed. Unfortunately, although neovastat can inhibit vascular growth in animal models, it has little or no benefit for cancer patients (Refs Reference Escudier215, Reference Lu216, Reference Loprinzi217). In summary, the above results indicate that the activation of NF-kB, especially in ECs, is probably an effective method for the treatment of cancer.
Conclusion
As a multi-effect transcription factor, NF-κB is involved in various biological processes such as inflammation, immune response, apoptosis and tumour. Stimulators that activate NF-κB signalling pathway include cytokines, chemokines, oxidative stress, bacterial LPS and so on. In the last few years, a large number of increasing research showed that NF-κB signalling molecules play an important role in regulating vascular-related diseases, especially tumours. In inflammation-related diseases, pathological angiogenesis is not only regulated by NIK-mediated non-canonical NF-κB signalling pathway, but also regulated by miR RNAs targeting to inhibit NF-κB-related mRNA expression. Moreover, the activation of NF-κB seems to have double-sided effects on tumourigenesis, progression and angiogenesis affecting tumour growth, invasion and migration. Based on the study of tumour growth and diffusion pathways, anti-angiogenesis treatment has become an important strategy that cannot be ignored. Although new insights have been provided into the biological function of NF-κB in angiogenesis and its role in some diseases such as cancer, there is still little work on NF-κB or IKK targeted drugs as angiogenesis regulators in clinical trials. More and more angiogenic inhibitors are required to explore the inhibition of tumour angiogenesis by regulating the NF-κB signalling pathway. In addition, the available literature to date mainly describes it as a tumour promotor, but this could not be the case in all tumour subtypes. These results indicate that more clinical samples need to be included in the study. Finally, the role of NF-κB pathway in tumour angiogenesis remains controversial, and whether NF-κB is used as a target or a medium for inhibiting angiogenesis needs to be further investigated.
Author contributions
R. M. contributed to research orientation, creativity and administrative support. Y. J., C. S. and X. L. contributed to the collection and collation of the literature. Y. J., J. Z. and C. S. contributed to the concept and design of this study and the editing of the manuscript. All authors have read and approved the final version of the manuscript.
Financial support
This work was supported by the National Natural Science Foundation of China (82070505); the Distinguished Professorship Program of Jiangsu Province to Renfang Mao; Jiangsu Postgraduate Research and Practice Innovation Project (KYCX22_3357); Nantong Science and Technology (JCZ2022072).
Competing interest
None.
Ethical standards
This study was approved by the ethics committee of Nantong University Medical School. Institutional approval was not required to publish this manuscript.






