1. Introduction
Microcephaly is a rare neurodevelopmental disorder defined by biparietal diameter (BPD) based on a Brazilian study where small head size is below 3–4 standard deviations and the cerebral cortex region of the brain is reduced in size (Araujo Junior et al., Reference Araujo Junior, Martins Santana, Martins, Junior, Ruano, Pires and Filho2014). Microcephaly patients may or may not have mild to severe mental retardation as well as squat physique and seizures or hereditary hearing loss (Darvish et al., Reference Darvish, Esmaeeli-Nieh, Monajemi, Mohseni, Ghasemi-Firouzabadi, Abedini, Bahman, Jamali, Azimi, Mojahedi, Dehghan, Shafeghati, Jankhah, Falah, Soltani, Banavandi, Ghani, Garshasbi, Rakhshani, Naghavi, Tzschach, Neitzel, Ropers, Kuss, Behjati, Kahrizi and Najmabadi2010). It can be classified into two forms; primary microcephaly (at birth) or secondary microcephaly (onset after birth) with and without other syndromic features (Cowie, Reference Cowie1960). Primary microcephaly is a prenatal developmental neurogenic disorder whereas secondary microcephaly is associated with progressive neurodegenerative disease.
Autosomal recessive primary microcephaly (MCPH) is a neurogenic mitotic disorder with normal neuronal migration, neuronal apoptosis and neural function of affected patients. There are various genetic and environmental causes including chromosomal aberrations, dented DNA as a consequence of incorrect mitotic spindle alignment, impulsive chromosomal abridgment, maternal overconsumption of alcohol, congenital infections, drugs taken during pregnancy, brain injury, metabolic disorders such as alaninuria or reaction to teratogenic remedies and substances taken during pregnancy (Darvish et al., Reference Darvish, Esmaeeli-Nieh, Monajemi, Mohseni, Ghasemi-Firouzabadi, Abedini, Bahman, Jamali, Azimi, Mojahedi, Dehghan, Shafeghati, Jankhah, Falah, Soltani, Banavandi, Ghani, Garshasbi, Rakhshani, Naghavi, Tzschach, Neitzel, Ropers, Kuss, Behjati, Kahrizi and Najmabadi2010; Faheem et al. Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015). Metabolic disorders often cause secondary rather than primary microcephaly and are often associated with additional symptoms and clinical signs (Von der Hagen et al., Reference von der Hagen, Pivarcsi, Liebe, von Bernuth, Didonato, Hennermann, Bührer, Wieczorek and Kaindl2014). Inquiries of metabolic screening, if necessary, must primarily focus on maternal phenylketonuria, phosphoglycerate dehydrogenase paucity and Amish fatal microcephaly (2-ketoglutaric aciduria) as a secondary basis of microcephaly (Kelley et al. Reference Kelley, Robinson, Puffenberger, Strauss and Morton2002). Rare metabolic causes of primary microcephaly include serine biosynthesis defects, sterol biosynthesis disorders, mitochondriopathies and congenital disorders of glycosylation (Von der Hagen et al., Reference von der Hagen, Pivarcsi, Liebe, von Bernuth, Didonato, Hennermann, Bührer, Wieczorek and Kaindl2014).
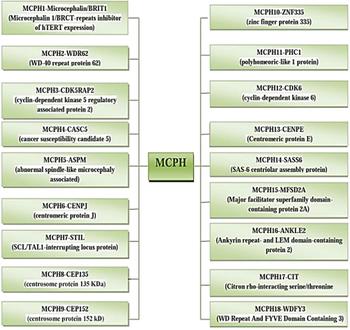
The incidence of MCPH ranges from 1:30,000–1:250,000 per live-birth depending on the population (Zaqout et al. Reference Zaqout, Morris-Rosendahl and Kaindl2017). In Asian and Arab populations where consanguineous marriages are mutual, MCPH is more common than in whites (Muhammad et al., Reference Muhammad, Mahmood Baig, Hansen, Sajid Hussain, Anjum Inayat, Aslam, Anver Qureshi, Toilat, Kirst, Wajid and Nürnberg2009). Worldwide MCPH has been reported in excess of 300 families and distinct patients; often with only sparse phenotype descriptions. Separate from intellectual infirmity (IQ between 30 and 70–80), hyperactivity and devotion deficit, dialog deferral, and a tapered slanting forehead, MCPH patients ordinarily do not have any auxiliary neurological ciphers (Passemard et al., Reference Passemard, Titomanlio, Elmaleh, Afenjar, Alessandri, Andria, de Villemeur, Boespflug-Tanguy, Burglen, Del Giudice and Guimiot2009; Kaindl et al., Reference Kaindl, Passemard, Kumar, Kraemer, Issa, Zwirner, Gerard, Verloes, Mani and Gressens2010; Bhat et al., Reference Bhat, Girimaji, Mohan, Arvinda, Singhmar, Duvvari and Kumar2011; Kraemer et al., Reference Kraemer, Picker-Minh, Abbasi, Fröhler, Ninnemann, Khan, Ali, Chen and Kaindl2016). To date, for MCPH 18 loci and residing genes are found to have mutations and are described in Figure 1, and their cytogenic locations are summarized in Figure 2 (Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015; Zaqout et al., Reference Zaqout, Morris-Rosendahl and Kaindl2017). The most common causes of MCPH are biallelic mutations in ASPM (68.6%), followed by those in the WDR62 gene (14.1%) and MCPH1 gene (8%). More genetic loci are still expected to exist given the lack of mutations in known loci in approximately 50–75% of western Europeans or North Americans with MCPH and approximately 20–30% of Indians or Pakistanis with MCPH (Verloes et al., Reference Verloes, Drunat, Gressens, Passemard, Pagon, Adam, Ardinger, Wallace, Amemiya, Bean, Bird, Ledbetter, Mefford, Smith and Stephens1993; Sajid Hussain et al., Reference Sajid Hussain, Marriam Bakhtiar, Farooq, Anjum, Janzen, Reza Toliat, Eiberg, Kjaer, Tommerup, Noegel, Nürnberg, Baig and Hansen2013).
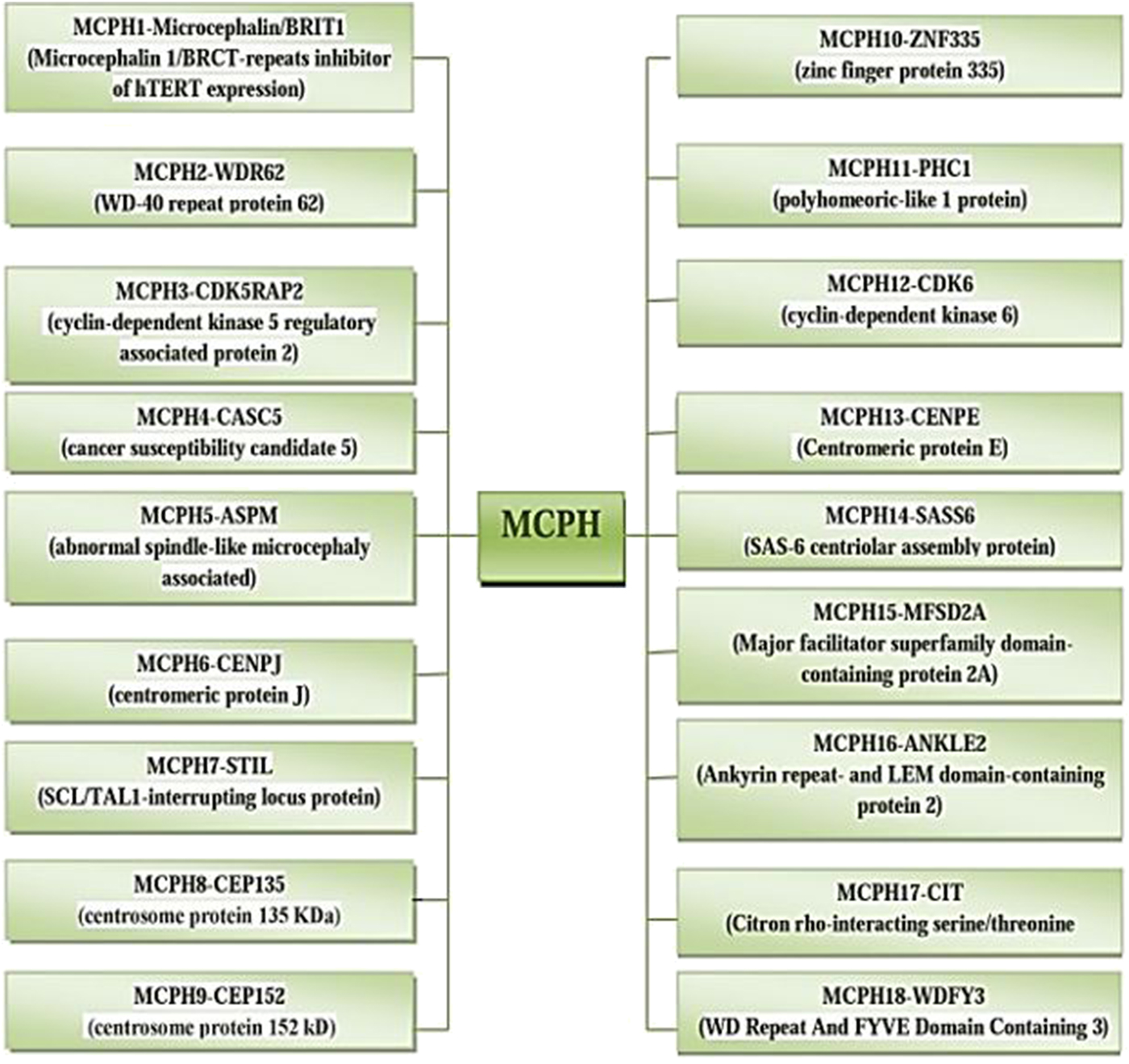
Fig. 1. MCPH loci and residing genes responsible for microcephaly.
Fig. 2. Cytogenic location of MCPH1–18 genes.
The up-to-date knowledge on the molecular genetics of MCPH, including the newly notorious locus with its gene (MCPH1–MCPH18), is expansively discussed in this review article. We review the corresponding genes and the proteins encoded by these genes, their probable role in the emerging brain (Table 1) and discuss mutations of these genes found in the Pakistani population (Table 2). In addition, the potential for these genes to perform various cognitive roles during human brain evolutionary processes is discussed.
Table 1. Functions of MCPH genes causing microcephaly.

AD: Autosomal dominant; AR: Autosomal recessive; MCPH: Autosomal recessive primary microcephaly.
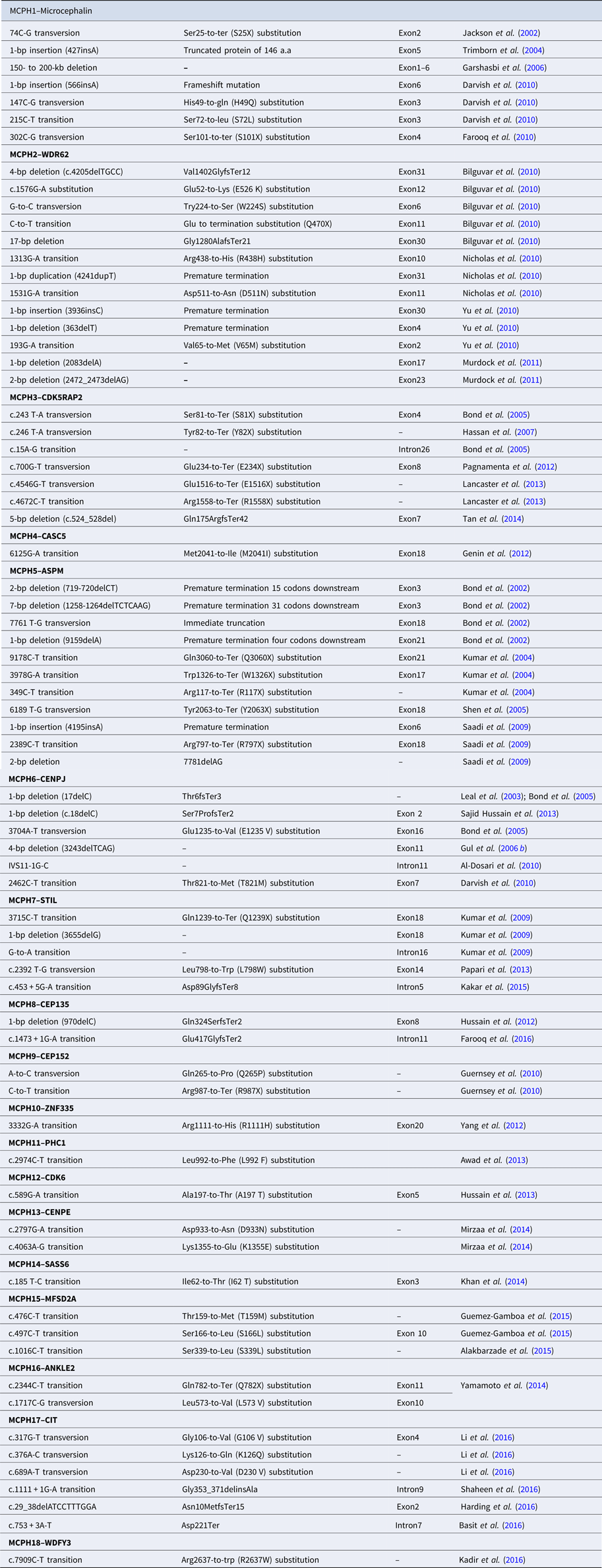
Table 2. Reported mutations of MCPH1–MCPH18 genes.
2. Molecular genetics
(i) MCPH1 (microcephalin)
MCPH1 encodes the important regulator of chromosome condensation (BRCT–BRCA1 C-terminus) the ‘Microcephalin protein’. The Microcephalin protein consists of three BCRT domains and conserved tandem repeats of phospho-peptide interacting amino acids (Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015; Pulvers et al., Reference Pulvers, Journiac, Arai and Nardelli2015). This gene is located on chromosome 8p23 comprising 14 exons, 835 amino acids, and with a genome size and molecular weight of 241,905 bp and 92,877 Da, respectively. It exists in three isoforms obtained after splicing and an open reading frame (ORF) of approximately 8032 bp (Venkatesh et al., Reference Venkatesh, Nagashri, Swamy, Mohiyuddin, Gopinath and Kumar2013; Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015). The Microcephalin protein, being a pleiotropic factor, imparts its significant effect in neurogenesis; it regulates the division of neuroprogenitor cells and prevents them from exhaustion, that is, from microcephaly. It is the significant regulator of telomere integrity and involved in the DNA damage repair mechanism. It also acts as a tumour suppressor in several human cancers, in germline functions and performs its function in brain development and in the regulation of cerebral cortex size (Venkatesh et al., Reference Venkatesh, Nagashri, Swamy, Mohiyuddin, Gopinath and Kumar2013; Pulvers et al., Reference Pulvers, Journiac, Arai and Nardelli2015). Some studies have reported that the MCPH1 gene is involved in brain size determination and as a positive selector for primate lineage (Montgomery et al., Reference Montgomery and Mundy2014). It was considered that MCPH1 may be a common denominator in the pathway of causing microcephaly enclosing the spectrum of both environmental and genetic causes. It leads to the drastic reduction of brain size, especially in the region of cerebral cortex, and short stature. Magnetic resonance imaging (MRI) of patients revealed the existence of cerebral deformations, such as the gyral pattern observed in the brain and corpus callosum hypoplasia. It is caused by premature switching of symmetric neuroprogenitors to asymmetric division. Mutation in MCPH1 causes mis-regulated chromosomal condensation, genomic instability and delayed de-condensation post mitosis (Liu et al., Reference Liu, Zhou and Wang2016). Successful experiments for MCPH1 mouse models reported mis-regulated mitotic chromosome condensation, deficiency in DNA repair, defective spindle orientation and a reduced skull size with approximately 20% reduction in body weight (Zhou et al., Reference Zhou, Tapias, Bruhn, Gruber, Sukchev and Wang2013; Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015).
(ii) MCPH2 (WDR62)
The WDR62 gene encodes the ‘WD repeat-containing protein 62’, located on chromosome 19q13.12, comprising 35 exons, 1518 amino acids, with a genomic size of 50,230 bp and a molecular mass of 165,954 Da, it has four known spliced isoforms (Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015; Pervaiz & Abbasi, Reference Pervaiz and Abbasi2016). WDR62 consists of a WD40 domain, a JNK docking domain and a MKK7 binding domain. It is a scaffold protein involved in the pathway of the c-Jun N-terminal kinase (Bastaki et al., Reference Bastaki, Mohamed, Nair, Saif, Tawfiq, Aithala, El-Halik, Al-Ali and Hamzeh2016) being highly expressed in neuronal precursors and within post mitotic neurons of the developing brain and in the ventricular and sub ventricular zone in the forebrain region (Pervaiz & Abbasi, Reference Pervaiz and Abbasi2016). This protein plays a significant part in the formation of several cellular layers in the cerebral cortex region during embryogenesis. It plays a compelling role in the proliferation and migration of neurons and in the duplication of centrioles that are dependent on mother centrioles (Sgourdou et al., Reference Sgourdou, Mishra-Gorur, Saotome, Henagariu, Tuysuz, Campos, Ishigame, Giannikou, Quon, Sestan, Caglayan, Gunel and Louvi2017). Several findings revealed that WDR62 functions are somehow similar to ASPM which is another MCPH gene. Moreover, studies revealed that the WDR62 gene, as it plays an important part in brain cortical development, is involved in human brain evolutions indicted by dramatic inflation in the size of the cerebral cortex (Pervaiz et al., Reference Pervaiz and Abbasi2016). Mutation in WDR62 can lead to a wide range of disorders including: microcephaly, cognitive disability, cortical malformations and multiple transcript variants by alternative splicing. The majority of mutations found in WDR62 revealed that these are responsible for approximately 10% of cases of microcephaly. Homozygous or heterozygous mutations in WDR62 both cause MCPH with or without cortical malformations (Naseer et al., Reference Naseer, Rasool, Sogaty, Chaudhary, Mansour, Chaudhary, Abuzenadah and Al-Qahtani2017). Patients with these mutations have a head circumference ranging from normal to severe. MRI of patients with these mutations showed numerous cortical malformations such as pachygyria, impulsivity, polymicrogyria, aggression, hypoplasia of corpus callosum, delayed psychomotor development, simplified gyral patterns, mental retardation with reduced head size and lissencephaly (Farag et al., Reference Farag, Froehler, Oexle, Ravindran, Schindler, Staab, Huebner, Kraemer, Chen and Kaindl2013; Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015; Bastaki et al., Reference Bastaki, Mohamed, Nair, Saif, Tawfiq, Aithala, El-Halik, Al-Ali and Hamzeh2016). Disruption of WDR62 in a mouse model altered the late neurogenesis neocortical progenitors proliferation process which further depicts asymmetric centrosome inheritance abnormalities leading to microcephaly in mice, and impaired mitotic cycle progression, causing temporary arrest at pro metaphase, as well as defects in spindle pole localization of WDR62 (Farag et al., Reference Farag, Froehler, Oexle, Ravindran, Schindler, Staab, Huebner, Kraemer, Chen and Kaindl2013; Sgourdou et al., Reference Sgourdou, Mishra-Gorur, Saotome, Henagariu, Tuysuz, Campos, Ishigame, Giannikou, Quon, Sestan, Caglayan, Gunel and Louvi2017).
(iii) MCPH3 (CDKRAP2)
The CDK5RAP2 gene is located on chromosome 9q33.2, comprising 39 exons, 1893 amino acids with a molecular mass of 215,038 Da (Graser et al., Reference Graser, Stierhof and Nigg2007). This gene is also known as C48, Cep215 and MCPH3. Cnn-1N is a small motif present at the N-terminal of a group of centrosome or spindle pole body associated proteins, such as Mto1 and Pcp1 from Schizosaccharomyces pombe, centrosomin from flies and CDK5RAP2 from mammals (Megraw et al., Reference Megraw, Li, Kao and Kaufman1999; Verde et al., Reference Verde, Pahlke, Salanova, Zhang, Wang, Coletti, Onuffer, Jin and Conti2001; Flory et al., Reference Flory, Morphew, Joseph, Means and Davis2002; Venkatram et al., Reference Venkatram, Tasto, Feoktistova, Jennings, Link and Gould2004; Conduit et al., Reference Conduit, Feng, Richens, Baumbach, Wainman, Bakshi, Dobbelaere, Johnson, Lea and Raff2014). The CDK5RAP2 gene encodes a regulator of CDK5 activity which is localized in the Golgi complex and centrosome in cells and in the cerebral cortex of the brain (Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015). It interacts with pericentrin and CDK5R1 and plays an important role in microtubule nucleation and centriole engagement. CDK5RAP2 being a part of the pericentriolar material is crucial for the microtubule organization capacity of the centrosome. Patients exhibiting the primary fibroblasts had a drastic reduction in the amount of CDKRAP2 and showed nuclear and centrosomal abnormalities and an increased frequency of alteration in cell size and migration. In addition, researchers also identified interplay of CDK5RAP2 with the constituents of the Hippo pathway, the transcriptional regulator TAZ and MASTI kinase. This finding enabled us to understand the mechanism of the Hippo pathway, revealing its role in the regulation of centrosome number. Higher levels of TAZ and Yup in fibroblast patients are observed but none of the other genes involved in the Hippo pathway have been seen to be downregulated. Modifications observed in the Hippo pathway constituents could consequently alter cellular properties and centrosomal deficiencies in patients with affected fibroblasts. This could further be relevant to MCPH controlling brain size and development (Sukumaran et al., Reference Sukumaran, Stumpf, Salamon, Ahmad, Bhattacharya, Fischer, Müller, Altmüller, Budde, Thiele and Tariq2017). Robustly connected with microtubules, centrosome and Golgi apparatus, CDK5RAP2 is mainly found inside the neural progenitors of the ventricular and sub ventricular areas of the developing brain; it has also been discovered in glial cells and early born neurons and is gradually downregulated when brain maturation occurs. In spindle checkpoint regulation CDK5RAP2 also imparts its effect. The damage to CDK5RAP2 shows chromosomal segregation and spindle checkpoint protein expression issues via binding in HeLa cells (Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015).
Scientist have determined that the ‘an’ homozygous mutation in Hertwig's anemia within the CDK5RAP2 gene results in deletion of exon four. Mutant mice exhibited microcephaly symptoms along with hypoplasia of brain regions – cortex and hippocampus – and hematopoietic phenotype. Mutant mice neuronal progenitors confirmed proliferative and survival defects and underwent apoptosis. The impaired centrosomal characteristic and altered mitotic spindle orientation in neuronal progenitors are consequences of CDK5RAP2 mutation (Lizarraga et al., Reference Lizarraga, Margossian, Harris, Campagna, Han, Blevins, Mudbhary, Barker, Walsh and Fleming2010).
(iv) MCPH4 (CASC5)
CASC5 is a protein coding gene, located on chromosome 15q15.1, comprising 27 exons, 2342 amino acids, with a molecular mass of 265 kDa. It is also known as hKNL-1, D40, Spc7, MCPH4, CT29, AF15Q14, PPP1R55 and hSpc105. In cells, the CASC5 gene is present in the nucleoplasm and in the brain the CASC5 gene is present in the cerebral cortex (Petrovic et al., Reference Petrovic, Mosalaganti, Keller, Mattiuzzo, Overlack, Krenn, De Antoni, Wohlgemuth, Cecatiello, Pasqualato and Raunser2014). The protein encoded by this gene is an important part of the multiprotein assembly that is used for the proper development of kinetochore–microtubule coupling and chromosome separation. It's a conserved scaffold protein that is used for proper kinetochore assembly, checkpoint functioning and spindle assembly. Unlike other MCPH proteins that are present around the centrosome, the CASC5 protein is used for coupling of chromatin with the mitotic apparatus and also interacts with BUB1, which properly manages the spindle assembly checkpoint. Any reduction in CASC5 protein causes chromosome misalignment and drives the cell into the mitosis (Saadi et al., Reference Saadi, Verny, Siquier-Pernet, Bole-Feysot, Nitschke, Munnich, Abada-Dendib, Chaouch, Abramowicz and Colleaux2016). Mutated CASC5 disrupts hMIS12, which is vital for correct chromosome alignment and segregation. Frequently this gene is part of the metaphase chromosome kinetochore and elevated mitotic index in patient cells designated mitotic arrest within the cells carrying the mutation. Lobulated and fragmented nuclei have also been identified in addition to micronuclei inside the affected cells. Moreover, altered DNA harms reactions with high peaks of γH2AX and 53BP1 seen in mutated cells as compared to control fibroblasts. These studies confirm the role of CASC5 in primary microcephaly (Szczepanski et al., Reference Szczepanski, Hussain, Sur, Altmüller, Thiele, Abdullah, Waseem, Moawia, Nürnberg, Noegel and Baig2016).
(v) MCPH5 (ASPM)
ASPM is located on chromosome 1q31.3, comprising 62,567 bp, 3477 amino acids, 28 exons divided into 10,906 ORFs (Saunders et al., Reference Saunders, Avides, Howard, Gonzalez and Glover1997; Ponting, Reference Ponting2006). It is present on spindle poles and centrosomes during mitosis. The domains of ASPM includes an N-terminus, an 81 IQ (isoleucine–glutamine) domain and a Calponin homology domain (Craig & Norbury, Reference Craig and Norbury1998; Bond et al., Reference Bond, Roberts, Mochida, Hampshire, Scott, Askham, Springell, Mahadevan, Crow, Markham and Walsh2002; Kouprina et al., Reference Kouprina, Pavlicek, Collins, Nakano, Noskov, Ohzeki, Mochida, Risinger, Goldsmith, Gunsior, Solomon, Gersch, Kim, Barrett, Walsh, Jurka, Masumoto and Larionov2005) and a C-terminus without any domain weighing about 220 kDa (Bond et al., Reference Bond, Scott, Hampshire, Springell, Corry, Abramowicz, Mochida, Hennekam, Maher, Fryns and Alswaid2003). The orthologs of ASPM include almost 20 different species. The cerebral cortex, the ganglia and the mouse neocortex show the expression of the gene during the process of neurogenesis (Bond et al., Reference Bond, Scott, Hampshire, Springell, Corry, Abramowicz, Mochida, Hennekam, Maher, Fryns and Alswaid2003; Paramasivam et al., Reference Paramasivam, Chang and LoTurco2007). ASPM downregulation is a result of neurosphere differentiation (Thornton & Woods, Reference Thornton and Woods2009). ASPM plays a role in pole organization through activation of kinesin-14 and CDK5RAP2 in normal cells (Tungadi et al., Reference Tungadi, Ito, Kiyomitsu and Goshima2017). The most important and crucial role of the ASPM gene is to perform cytokinesis during meiosis. ASPM plays a fundamental role in organizing microtubules and focusing spindle poles during mitosis (do Carmo Avides & Glover, Reference do Carmo Avides and Glover1999). ASPM knockdown mediated by Morpholino results in the reduction of head size in different species such as Zebra fish (Kim et al., Reference Kim, Lee, Choi, Jung, Ahn, Yeo, Choi and Kim2011). The reduced surface area and increased thickness of white matter are consequences of loss of ASPM, while preserving the memory of patients in contrast to its intellectual disabilities (Passemard et al., Reference Passemard, Verloes, de Villemeur, Boespflug-Tanguy, Hernandez, Laurent, Isidor, Alberti, Pouvreau, Drunat and Gérard2016). ASPM knockdown effects could be reduced by CITK overexpression and CITK microcephaly phenotype is a result of spindle orientation (Gai et al., Reference Gai, Bianchi, Vagnoni, Vernì, Bonaccorsi, Pasquero, Berto, Sgrò, Chiotto, Annaratone and Sapino2016). The similarities of WDR62 and ASPM includes the same location and physical interaction during interphase mediated by CEP63. Loss of WDR62 and ASPM in the developing brain induces shortage of cilia and centrosomes. Both genes determine the fate of cells and localize CPAP to the centrosome (Jayaraman et al., Reference Jayaraman, Kodani, Gonzalez, Mancias, Mochida, Vagnoni, Harper, Reiter, Yu, Bae and Walsh2017). Different types of mutations have been observed in the ASPM gene in 33 families from Pakistan (Gul et al., Reference Gul, Hassan, Mahmood, Chen, Rahmani, Naseer, Dellefave, Muhammad, Rafiq, Ansar, Chishti, Ali, Siddique and Ahmad2006 a). These mutation types include deletion, substitution, duplication and variation in the intrinsic region (Muhammad et al., Reference Muhammad, Mahmood Baig, Hansen, Sajid Hussain, Anjum Inayat, Aslam, Anver Qureshi, Toilat, Kirst, Wajid and Nürnberg2009; Saadi et al., Reference Saadi, Borck, Boddaert, Chekkour, Imessaoudene, Munnich, Colleaux and Chaouch2009). The ASP gene in mutant form was first discovered in Drosophila (do Carmo Avides & Glover, Reference do Carmo Avides and Glover1999). Different experiments were performed to study mutations in ASPM. One study examined the developing cerebral cortex in two mutant mouse lines and concluded that the reduced size of the brain was a consequence of ASPM mutation (Riparbelli et al., Reference Riparbelli, Callaini, Glover and Avides2002). Using whole-genome sequencing, a mutation in exon 16 in the ASPM gene was seen, and non-syndromic microcephaly with altered IQ number was reported.
(vi) MCPH6 (CENPJ)
CENPJ is present on chromosome 13q12.2, is comprised of 40,672 bp, 1338 amino acids, 17 exons distributed in 5187 bp ORFs, weighing approximately 153 kDa (Saunders et al., Reference Saunders, Avides, Howard, Gonzalez and Glover1997). The domains of CENPJ include protein phosphorylation domains, five coiled-coil domains (CCDs), and the C-terminal domain has 21 G-box repeats and a leucine zipper motif (Hung et al., Reference Hung, Tang and Tang2000; Bond et al., Reference Bond, Roberts, Springell, Lizarraga, Scott, Higgins, Hampshire, Morrison, Leal, Silva and Costa2005). The gene holds its position within the centriole. The CENPJ gene has almost 204 orthologs. CENPJ carries out microtubule assembly in the centrosome by causing nucleation and depolymerization of microtubules (Hung et al., Reference Hung, Chen, Chang, Li and Tang2004). CENPJ also maintains centriole integrity with rearrangement of microtubules (Kirkham et al., Reference Kirkham, Müller-Reichert, Oegema, Grill and Hyman2003). During neurogenesis, the frontal cortex neuro-epithelium expresses the CENPJ gene (Bond et al., Reference Bond, Roberts, Springell, Lizarraga, Scott, Higgins, Hampshire, Morrison, Leal, Silva and Costa2005). Damage to the structure of the centrosome causes cell arrest in mitosis and is caused by low levels of the CENPJ protein (Cho et al., Reference Cho, Chang, Chen and Tang2006). CENPJ knockdown increases the rate of multiple spindle poles, apoptosis and mitosis arrest and loss of centrioles in Drosophila (Koyanagi et al., Reference Koyanagi, Hijikata, Watashi, Masui and Shimotohno2005; Basto et al., Reference Basto, Lau, Vinogradova, Gardiol, Woods, Khodjakov and Raff2006). Drosophila flies without centrioles die at an early age due to loss of cilia or flagella (Stevens et al., Reference Stevens, Raposo, Basto, St Johnston and Raff2007). The total number of mutations in the CENPJ gene never exceeded five. The first mutation was discovered in a Brazilian family, the second and third mutations belonged to a Pakistani family (Bond et al., Reference Bond, Roberts, Springell, Lizarraga, Scott, Higgins, Hampshire, Morrison, Leal, Silva and Costa2005) and the forth was discovered in people suffering from Seckel syndrome (Al-Dosari et al., Reference Al-Dosari, Shaheen, Colak and Alkuraya2010). Studies showed that in the third mutation, four consecutive nucleotide units (TCAG) are deleted from around 19 bp downstream of exon 11 resulting in frameshifting and premature termination of protein (Gul et al., Reference Gul, Hassan, Hussain, Raza, Chishti and Ahmad2006b).
(vii) MCPH7 (STIL)
STIL is located on chromosome 1p33, comprising 63,018 bp with an ORF of 5225 bp, 20 exons, 1287 amino acids and a molecular weight of 150 kDa (Kumar et al., Reference Kumar, Girimaji, Duvvari and Blanton2009; Kaindl et al., Reference Kaindl, Passemard, Kumar, Kraemer, Issa, Zwirner, Gerard, Verloes, Mani and Gressens2010). It has recently been reported that the human oncogene SCL/TAL1 locus is thoroughly conserved in vertebrate species, and is involved in regulation of toxic susceptibility in PC12 cells through the sonic hedgehog (Shh) pathway. Knockdown of STIL expression by RNAi showed no effect on survival of proliferating PC12 cells following increased cell death in differentiated neurons after drug analysis. In case of overexpression of STIL in proliferating cells toxic susceptibility is increased but it causes no effect in mature neurons (Szczepanski et al., Reference Szczepanski, Hussain, Sur, Altmüller, Thiele, Abdullah, Waseem, Moawia, Nürnberg, Noegel and Baig2016). Biochemical, cell biology and biophysical analysis reported that STIL retains a central short CCD, which suggests a critical role in oligomerization, centrosomal localization and protein interaction. Protein interaction is mediated by the central intrinsically disordered region of STIL, comprising 400–700 residues, just like CPAP interaction during centriole duplication. Generally, STIL is a disordered protein retaining three structured regions: N-terminal domain possessing 1–370 residues, 1062–1148 residues identified as a STIL ANA-2 motif and a short segment of 718–750 that give rise to a CCD. Size exclusion chromatography suggested that the CCD peptide comprises α helices that form a quaternary structure. Repeating patterns of hydrophobic, hydrophilic and charged residues present at specified locations to form a CCD. Recent studies revealed that two regions retaining eight hydrophobic residues exist in this domain and that these regions mediate oligomerization of CCD. Within this domain hydrophobic residues form axes occupied by hydrophilic residues promoting hydrophobic interactions that are an important part of protein structure and stability. Two leucine residues, L718 and L736, identified in this hydrophobic region lead to oligomerization of the CCD, and L736 has a greater role in this process. Further inspection revealed that mutations in these residues did not affect the secondary structure but alter the quaternary configuration which leads to malfunction of STIL. The CCD has a key role in oligomerization and promoting STIL function in self-interaction, centriolar replication, embryogenesis and in development of cilia. Current studies report that interaction of one helix of STIL with a PLK4 PB3 domain occurs via hydrophobic residues in the CCD and promotes centriole duplication. Phosphorylation of STIL by PLK4 facilitates STIL and SAS-6 protein interaction. Note that the L736 residue is critically involved in the interaction with PLK4. The hydrophobic core in the CCD of STIL imparts oligomerization building dimers and tetramers, which are analogues of its Drosophila ortholog Ana-2 and Caenorhabditis elegans centrosomal protein SAS-6 (David et al., Reference David, Amartely, Rabinowicz, Shamir, Friedler and Izraeli2016). Clinical and molecular genetic studies revealed that MCPH is rarely caused by STIL mutation (2.2%). The expression study demonstrated that STIL is critically involved in early forebrain development that may be associated with the Shh signalling pathway (Mouden et al., Reference Mouden, de Tayrac, Dubourg, Rose, Carre, Hamdi-Roze, Babron, Akloul, Héron-Longe, Odent, Dupé, Giet and David2015).
(viii) MCPH8 (CEP135)
The CEP135 gene is located on chromosome 4q12, comprising 26 exons and 1140 amino acids, it encodes a centrosomal protein that is a reserve helical protein detected throughout the cell cycle at the centrosomes giving greater strength to the centrosomes. Recent studies revealed that CEP135 in Drosophila plays a crucial role in central microtubule pair assembly in sperm axoneme and asymmetric cell division of neuroblasts. Intriguingly, in Homo sapiens and Drosophila microtubule binding sites have been mapped to the N-terminal. Fluorescence microscopy, cryo-electron analysis, including biochemical analysis, elucidated that in vitro development of microtubule bundles is induced by the interaction of CEP135 with tubulin, protofilaments and microtubules. A microtubule binding site has been detected between 96–108 residues and this segment integrated with positively charged surface patch 2; this was discovered in an atomic CEP135 model study, which suggested that the basic amino acids of the microtubule binding domain mediate interactions with the outer surface of the microtubule that is negatively charged (Hilbert et al., Reference Hilbert, Noga, Frey, Hamel, Guichard, Kraatz, Pfreundschuh, Hosner, Flückiger, Jaussi and Wieser2016). Further studies of major microtubule binding sites discovered a segment of 13 amino acids comprising 96–108 residues leading to microtubule binding activity of CEP135-N. Within this segment three lysine residues, K101, K104 and K108, were identified that contribute highly positive electrostatic surface potential of patch 2. These three lysine residues play a critical role in efficient microtubule bundling or cross linking by CEP135-N. Binding ability of two or more microtubules of CEP135-N leads to the development of microtubule triplets or joining of adjacent triplets within the microtubule wall. In such a way, the centrosomal protein CEP135 plays a key role in the biosynthesis of centrosomes which control the cell (Hilbert et al., Reference Hilbert, Noga, Frey, Hamel, Guichard, Kraatz, Pfreundschuh, Hosner, Flückiger, Jaussi and Wieser2016).
(ix) MCPH9 (CEP152)
CEP152 is a protein coding gene located on chromosome 15q21.1, comprising 149,368 bp, 38 exons, 1710 amino acids, with a molecular mass of 195,626 Da (Jamieson et al., Reference Jamieson, Fryns, Jacobs, Matthijs and Abramowicz2000). The CEP152 gene is also known as Asterless, KIAA0912, SCKL5 and MCPH9 (Dzhindzhev et al., Reference Dzhindzhev, Quan, Weiskopf, Tzolovsky, Cunha-Ferreira, Riparbelli, Rodrigues-Martins and Bettencourt-Dias2010). In cells this gene is localized in centrosomes and in the brain it is localized in the cerebral cortex (Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015). Centrosomal protein 152 contains (KW-0175) a CCD (KW-9994), also known as a Heptad Repeat pattern including five different conserved protein sequences that lead to the coiled-coil protein domain formation, these are formed of 234–490, 615–664, 700–772, 902–993 and 1170–1241 amino acids (Kalay et al., Reference Kalay, Yigit, Aslan, Brown, Pohl, Bicknell, Kayserili, Li, Tüysüz, Nürnberg, Kiess, Koegl, Baessmann, Buruk, Toraman, Kayipmaz, Kul, Ikbal, Turner, Taylor, Aerts, Scott, Milstein, Dollfus, Wieczorek, Brunner, Hurles, Jackson, Rauch, Nürnberg, Karagüzel and Wollnik2011). The protein of this gene organizes the microtubules of cells and has a very important role in shaping the cell, polarity, receptivity and cellular division. The protein localized in the microtubule and centrosome interacts with PLK4, CENPJ (via N-terminus), CINP, CDKRAP2, WDR62, CEP63, CEP131 and DEUP1 (Cizmecioglu et al., Reference Cizmecioglu, Arnold, Bahtz, Settele, Ehret, Haselmann-Weiß, Antony and Hoffmann2010; Dzhindzhev et al., Reference Dzhindzhev, Quan, Weiskopf, Tzolovsky, Cunha-Ferreira, Riparbelli, Rodrigues-Martins and Bettencourt-Dias2010; Kalay et al., Reference Kalay, Yigit, Aslan, Brown, Pohl, Bicknell, Kayserili, Li, Tüysüz, Nürnberg, Kiess, Koegl, Baessmann, Buruk, Toraman, Kayipmaz, Kul, Ikbal, Turner, Taylor, Aerts, Scott, Milstein, Dollfus, Wieczorek, Brunner, Hurles, Jackson, Rauch, Nürnberg, Karagüzel and Wollnik2011; Fırat-Karalar & Stearns, Reference Fırat-Karalar and Stearns2014). The interaction of CEP152 with CEP63, CDK5RAP2 and WDR62 form a bit by bit assembled complex at the centrosome creating a ring close to the parental centriole. CEP152 plays an important role in centrosome duplication/shape and cell/polarity/motility, and also functions as a molecular scaffold that facilitates the interaction of CENPJ and PLK4, two molecules that play an important role in centriole configuration (Cizmecioglu et al., Reference Cizmecioglu, Arnold, Bahtz, Settele, Ehret, Haselmann-Weiß, Antony and Hoffmann2010). It is suggested to take PLK4 away from PLK4:CEP92 complexes in early G1 daughter centrioles and to reposition PLK4 at the outer boundary of a new forming CEP152 ring structure. It also plays an essential role in deutrosome-mediated centriole amplification that can form many centrioles. Overexpression of CEP152 can drive amplification of centrioles (Dzhindzhev et al., Reference Dzhindzhev, Quan, Weiskopf, Tzolovsky, Cunha-Ferreira, Riparbelli, Rodrigues-Martins and Bettencourt-Dias2010). The CEP152 (human) gene is an ortholog of the Drosophila asterless (asl) gene, comprising 72,835 bp and 1710 amino acids and with a protein with a molecular weight of 152 kDa. A mutation in the CEP152 protein that converts glutamine into proline is assumed to be pathogenic and can disrupt the potential coiled-coiled protein domain, which results in reduction of head size. Similarly, a greater head size reduction was also seen in compound heterozygous females compared with missense homozygous females. The truncated protein suggests a nonsense-mediated decay of the mutated transcript; these findings have also shown that throughout a functional assay to determine subcellular localization that the wild-type CEP152–GFP fusion protein can be found in γ-tubulin co-structures. Mutant CEP152 flagged with GFP failed to co-localize with the γ-tubulin, which furthermore substantiate pathogenicity caused by this mutation (Guernsey et al., Reference Guernsey, Jiang, Hussin, Arnold, Bouyakdan, Perry, Babineau-Sturk, Beis, Dumas, Evans and Ferguson2010).
(x) MCPH10 (ZNF335)
ZNF335 is a protein coding gene located on chromosome 20q13.12, comprising 24,258 bp, 28 exons, 1342 amino acids, with molecular mass of 144,893 Da (Deloukas et al., Reference Deloukas, Matthews, Ashurst, Burton, Gilbert, Jones, Stavrides, Almeida, Babbage, Bagguley and Bailey2001). The ZNF335 gene is also known as NIF-1, NIF-2 and MCPH10 (Mahajan et al., Reference Mahajan, Murray and Samuels2002; Garapaty et al., Reference Garapaty, Mahajan and Samuels2008). In the cell this gene is localized in the nucleus and in the brain it is localized in the cerebral cortex (Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015). ZNF335 codes for the zinc finger protein 335 and has a repeat domain and a zinc finger C2H2-type (IPR013087) domain (Klug, Reference Klug1999). The ZNF335 protein is a part or an associated component of some histone methyltransferase complexes and is also involved in regulating transcription through recruitment of these complexes to gene promoters and via the nuclear hormone receptor it also increases ligand dependent transcriptional activation. In addition, it is also very important for proliferation and self-renewal of neural progenitor cells by controlling the regulation of specific genes that are involved in brain development, including REST, and it is also involved in regulating the expression of genes that are involved in somatic development, for example, lymphoblast proliferation. This gene also encodes a component of the vertebrate-specific trithorax H3K4-methylation chromatin remodelling complex that controls central nervous system (CNS) gene expression and cell fate (Yang et al., Reference Yang, Baltus, Mathew, Murphy, Evrony, Gonzalez, Wang, Marshall-Walker, Barry, Murn and Tatarakis2012). ZNF335 is very important for progenitor cell division/differentiation; mutation in this gene leads to the degeneration of neurons, and knockdown of this gene causes a reduction in the size of the brain with a lack of cortex formation as well as disrupted differentiation and proliferation of neuronal cells (McLean et al., Reference McLean, Bhattarai, Hughes, Mahalingam and Bagasra2017). Due to a mutation in ZNF335, MCPH10 was identified in an affected member of an Arab Israeli family. The mutation was identified in exon 20 as a homozygous 3332G-A transition, which resulted in an Arg1111 to His (R1111H) substitution at the conserved residue in the 13th zinc-finger domain. This mutation was found at the final point of a splice donor site that disrupts normal splicing, which leads to the formation of unusually large transcripts that contain intron 19 and 20 with a proposed premature termination sequence (Yang et al., Reference Yang, Baltus, Mathew, Murphy, Evrony, Gonzalez, Wang, Marshall-Walker, Barry, Murn and Tatarakis2012). Levels of ZNF335 protein is severely reduced in patient cells. Various studies regarding this gene have revealed that ZNF335 is attached to the chromatin remodelling complex (analogue to the TrxG (trithorax) complex in Drosophila) involving H3K4 methyltransferase, which can control the expression of important genes in various pathways. nBAF is a neural-specific chromatin regulatory complex, any mutation in this complex disrupts the proliferation of cells, indicating that it can also lead to microcephaly. Deficiency of the ZNF335 genes leads to the degeneration of neurons, which makes it much more critical in contrast to the other microcephaly syndromes that are related to postnatal survival. Studies of in vitro and in vivo mouse models revealed that the knockdown of ZNF335 causes a severe reduction in the size of the brain, which has no cortex, and disturbs the differentiation and proliferation of nerve cells (Yang et al., Reference Yang, Baltus, Mathew, Murphy, Evrony, Gonzalez, Wang, Marshall-Walker, Barry, Murn and Tatarakis2012).
(xi) MCPH11 (PHC1)
The PHC1 gene is located on chromosome12p13.31 and encodes a protein containing 1004 amino acids with a molecular mass of 105,534 Da. The PHC1 gene is also known as Early Developmental Regulator 1, HPH1, EDR1, RAE28, PH1 and MCPH11 (Chavali et al., Reference Chavali, Stojic, Meredith, Joseph, Nahorski, Sanford, Sweeney, Krishna, Hosmillo, Firth and Bayliss2017). MCPH11 is a disease that is caused by a homozygous mutation in the PHC1 gene on chromosome 12p13. PHC1 takes part in the regulation of the cell cycle, and its mutation highlights the role of chromatin remodelling in the pathogenesis of primary microcephaly (Awad et al., Reference Awad, Al-Dosari, Al-Yacoub, Colak, Salih, Alkuraya and Poizat2013). Enhanced geminin expression was observed in cells with a PHC1 mutation. A core component of canonical PRC1 is PHC1 as a genetic basis for MCPH11 in a family with reported consanguinous homozygous missense PHC1 variants. The functional analysis of this pathogenic variant in patient cells revealed lower PHC1 expression with lower genome wide H2AUb1 levels and impaired recruitment of PHC1 to loci of DNA damage and repair (Srivastava et al., Reference Srivastava, McGrath and Bielas2017). Interestingly, the MCPH11 PHC1 variant essentially had an impact on neural development in affected individuals, potentially highlighting a sensitivity of the developing brain to dysregulation of H2AUb1 modification exchange. As PHC1 regulates the cell cycle, PHC1 mutation highlights the role of chromatin remodelling in the pathogenesis of primary microcephaly (Srivastava et al., Reference Srivastava, McGrath and Bielas2017; Triglia et al., Reference Triglia, Rito and Pombo2017).
(xii) MCPH12 (CDK6)
CDK6 encodes a protein of the cyclin dependent protein kinase (CDK) family, located on chromosome 7q21.2 with a genome size of 231,707 bp, 326 amino acids, 10 exons and a molecular weight of 36,983 Da. The CDK6 protein controls the mechanism of the cell cycle and plays a very significant role in differentiation of different cell types. A mutated CDK6 gene or knockdown cells cause distorted nuclei disorganization of the spindles and microtubule and supernumerary centrosomes, whereas a decrease in proliferation was also observed in patients. Mutation in cells also disturbs apical neuronal precursor cell proliferation, which leads to the imbalance between symmetric and asymmetric cell division that causes progenitor cell depletion that could be the basis for reduction in neuronal cell production that finally leads to primary microcephaly (Hussain et al., Reference Hussain, Baig, Neumann, Peche, Szczepanski, Nurnberg, Tariq, Jameel, Khan, Fatima, Malik, Ahmad, Altmüller, Frommolt, Thiele, Höhne, Yigit, Wollnik, Neubauer, Nürnberg and Noegel2013). In a recent study, most related genes that code for CDK6 were found to have a homozygous single nucleotide substitution, 589G > A. This mutation leads to altered mitosis because during the mitosis process patient primary fibroblasts failed to recruit CDK6 to the centrosome and it also affects cellular localization (Grossel et al., Reference Grossel, Baker and Hinds1999). Moreover, this study was also substantiated by another study that revealed reduced proliferation capacity of CDK6 patient primary fibroblasts and knockdown cells (Grossel & Hinds, Reference Grossel and Hinds2006).
(xiii) MCPH13 (CENPE)
CENPE is a protein coding gene located on chromosome 4q24, comprising 492,604 bp and 2701 amino acids. This gene encodes a protein that has signal transducer activity and sequence specific DNA binding activity. It also has transcription factor activity. Domains of this gene include Ploop_NTPase, Kinesin_motor_dom, CENPE, Kinesin_motor_CS and Kinesin-like FAM. MCPH is a heterogeneous autosomal recessive disorder. In last four years studies there has been increased knowledge about the new mutated genes involved in MCPH and intense work is done at both the clinical and cellular level to determine disease mechanisms. The functions of proteins encoded by WDR62, CASC5, PHC1, CDK6, CENP-E, CENP-F, CEP63, ZNF335, PLK4 and TUBGPC genes, have been added to the complex network of critical cellular processes that play an important role in brain growth and size. In a male child with the mutated CENPE gene and who had microcephaly, his head was seen to start squeezing at the age of 5 and after some years he died. The boy also had dysmorphic facial features, including, prominent nose and sloping forehead. His sister of 3 years old also had microcephaly and also showed the disease phenotype. Up until now various mutations have been found in CENPE (Ahmad et al., Reference Ahmad, Baig, Abdulkareem, Hussain, Sur, Toliat, Nürnberg, Dalibor, Moawia, Waseem and Asif2017).
(xiv) MCPH14 (SASS6)
The SAS-6 gene is located on chromosome-1p21.2, comprising 49,552 bp, 657 amino acids, 17 exons and a mass of 74,397 Da. The SAS-6 gene encodes a protein known as Spindle assembly abnormal protein-6 homolog, which is a coiled-coil protein. The gene is found within the cytoplasm, cytoskeleton, microtubule organizing centre, centrosome, cytoskeleton and centrioles. The most critical and fundamental role of the SAS-6 gene is to help in centriole formation by producing a procentriole. Centrioles play a crucial role in cilia and flagella and so are of much importance and their formation is strongly linked to cell duplication and centrosome replication (Gupta et al., Reference Gupta, Badarudeen, George, Thomas, Gireesh and Manna2015; Arquint & Nigg, Reference Arquint and Nigg2016). To ensure genome integrity, centriole replication is essential. SAS-6 self-assembles into a cartwheel structure, and is able to undergo dimerization and oligomerization thus supporting centriole formation. The most primitive pathway for assembling centrioles involves seven major constituents: STIL, γ-tubulin, Plk4, CPAP, Cep135, Cep135 and hSAS6.
After attachment of P1k4, Cep135 assembles at the parental centriole position, then phosphorylated STIL and hsSAS-6 gather and through a positive feedback mechanism form a complex with CPAP and initiate the formation of the procentriole (Ohta et al., Reference Ohta, Essner, Ryu, Palazzo, Uetake and Kuriyama2002; Gopalakrishnan et al., Reference Gopalakrishnan, Guichard, Smith, Schwarz, Agard, Marco and Avidor-Reiss2010). HsSAS-6 located in the cytoplasm gathers around the centriole and forms a complex with Cep135 to initiate cartwheel structure organization by oligomerization by passing through G1/S phase. G2/S phase leads to the stability of hsSAS-6 (Strnad et al., Reference Strnad, Leidel, Vinogradova, Euteneuer, Khodjakov and Gonczy2007; Keller et al., Reference Keller, Orpinell, Olivier, Wachsmuth, Mahen, Wyss, Hachet, Ellenberg, Manley and Gonczy2014). A family was selected from Dera Ismail Khan, an urban area in the province of Pakistan, Khyber Pakhtunkhwa. A pedigree analysis of five generations provided a result of four persons diagnosed with several physiological and mental issues resulting from consanguineous marriages. Out of four, two were girls, aged 3.5 and 5 years, and two were men, aged 42 and 50. All four experienced mental retardation, low level IQ (between 20–40), pronunciation issues and even walking problems. By performing several tests including tomography, STR, SNP, genome-wide linkage analysis and whole-genome analysis etc., it was concluded that the SAS-6 gene was responsible for this condition, which was determined to be primary microcephaly. In the PISA domain of the SAS-6 gene, the replacement of threonine with isoleucine at position 62 causes primary microcephaly. Microcephaly linked with hsSAS-6 is an autosomal recessive disease. The drastic impact of hsSAS-6 deficiency on centriole formation also effects cell division thus disturbing normal neurogenesis and ultimately affecting brain nourishment (Khan et al., Reference Khan, Rupp, Orpinell, Hussain, Altmüller, Steinmetz, Enzinger, Thiele, Höhne, Nürnberg and Baig2014).
(xv) MCPH15 (MFSD2A)
MFSD2A, also known as MCPH15 and NLS1, encodes a transmembrane protein required for brain uptake of omega-3 (Docosahexaenoic acid) and different long-chain fatty acids (lysophosphatidylcholine) (Alakbarzade et al., Reference Alakbarzade, Hameed, Quek, Chioza, Baple, Cazenave-Gassiot, Nguyen, Wenk, Ahmad, Sreekantan-Nair and Weedon2015; Guemez-Gamboa et al., Reference Guemez-Gamboa, Nguyen, Yang, Zaki, Kara, Ben-Omran, Akizu, Rosti, Rosti, Scott and Schroth2015). This gene is localized on chromosome 1p34.2, comprising 14 exons, 543 amino acids, with a genome size of 14,857 bp and a molecular weight of 60,170 Da. MFSD2A is mainly localized to the cytosol, plasma membrane and cytoplasmic bodies in the human brain as well as other cells. The MFSD2A gene is also present in the cerebral cortex (endothelial cells, neuronal cells, etc) and is an essential component of the blood–brain barrier (Reiling et al., Reference Reiling, Clish, Carette, Varadarajan, Brummelkamp and Sabatini2011). MFSD2A is evolutionarily conserved from teleost fish to humans. Mutation in the MFSD2A gene causes microcephaly leading to a brain volume that is smaller than usual. High expression of MFSD2A is observed in the blood–brain barrier of both humans and mice. Reduced brain DHA levels along with reduced LPC uptake and microcephaly as the most prominent phenotype is reported in MFSD2A-deficient mice (Nguyen et al., Reference Nguyen, Ma, Shui, Wong, Cazenave-Gassiot, Zhang, Wenk, Goh and Silver2014). As supported by these studies, LPC is essential for normal development of human and mice brains (Quek et al., Reference Quek, Nguyen, Fan and Silver2016). Most affected people have delayed speech and language skills, and delayed motor skills (standing, sitting and walking). Slim, sloping forehead, behavioural issues and short stature compared to others in their family was also observed. Two distinctive homozygous missense mutations were seen to occur in the MFSD2A gene in affected individuals from two consanguineous families from northern Africa with autosomal recessive microcephaly causing early death (Guemez-Gamboa et al., Reference Guemez-Gamboa, Nguyen, Yang, Zaki, Kara, Ben-Omran, Akizu, Rosti, Rosti, Scott and Schroth2015). Homozygous missense mutations occurring in the MFSD2A gene were also seen in affected individuals belonging to a consanguineous Pakistani family. It was also seen that morpholino knockdown of orthologous MFSD2A in Zebra fish brought about early postnatal lethality, microcephaly and blood–brain barrier interruption. Likewise, it was also found that MFSD2A-null mice had a roughly 40% expansion in plasma levels of LPC compared to controls with debilitated take-up of LPC into the brain (cerebrum) (Guemez-Gamboa et al., Reference Guemez-Gamboa, Nguyen, Yang, Zaki, Kara, Ben-Omran, Akizu, Rosti, Rosti, Scott and Schroth2015).
(xvi) MCPH16 (ANKLE2)
Several mutated genes have been identified to play an important role in the regulation of the cell cycle and cell proliferation. One of them is the ANKLE gene encoding the LEM4 protein, which plays a critical role in nuclear envelope formation through the mitotic phosphorylation of BAF protein during mitosis. Upon mitotic entry, the LEM protein interacts with a chromatin-binding protein known as Barrier to Autointegration. BAF regulates chromatin structure and chromosome segregation in gene expression and development. Recent studies have revealed that the ANKLE2 gene mapped to chromosome 12q24.33, has a size of 938 amino acids and a molecular mass of approximately 10,000 Da. According to current studies its N-terminal tail is followed by a transmembrane domain of approximately 40 amino acids, which is known as the LEM domain and retains two central ankyrin repeats and a cytoplasmic C-terminus. Mutation in this gene leads to abnormal development of the nuclear envelope during mitosis. Mutated ANKLE2 gene leads to impaired brain development and reduces brain size as a result of a decreased number of neuroblasts, reduced mitosis, less cell proliferation and increased apoptosis (Faheem et al., Reference Faheem, Naseer, Rasool, Chaudhary, Kumosani, Ilyas, Pushparaj, Ahmed, Algahtani, Al-Qahtani and Jamal2015). This improper development of the brain contributes to MCPH16, which is predominantly caused by consanguineous marriages. Clinical review or neuroimaging of patients with MCPH16 reveals symptoms such as reduced brain size, intellectual disability, knee contractures, enlarged posterior horns of the lateral ventricles, adducted thumbs and epilepsy (von der Hagen et al., Reference von der Hagen, Pivarcsi, Liebe, von Bernuth, Didonato, Hennermann, Bührer, Wieczorek and Kaindl2014). Recently, whole-exome sequencing and analysis of genes that are potentially involved in neurological disorders identified compound heterozygous mutations in the ANKLE2 gene in exon 11 (c.23344C-T transition as a result of Gln782 to Ter (Q782X) substitution), and in exon 10 (c.1717C-T as a result of Leu 573 to Val (L573 V) substitution). Analyses of brains from Drosophila identified a mutation in l (l) G0222, the homolog of the ANKLE2 gene in humans. This mutation results in loss of thoracic bristles and impairs development of sensory organs in clones. Such evidence suggests that genes identified in Drosophila clones and variant alleles in Homo sapien homologs in families with Mendelian diseases play a critical role in the discovery of disease causing genes and the mechanisms involved in these disorders (Yamamoto et al., Reference Yamamoto, Jaiswal, Charng, Gambin, Karaca, Mirzaa, Wiszniewski, Sandoval, Haelterman, Xiong and Zhang2014).
(xvii) MCPH17 (CIT)
The CIT gene encoding citron rho-interacting serine/threonine kinase is critical for the development of a normal sized brain. CIT has an N-terminal kinase domain and many C-terminal domains that aid interaction between the components of its contractile ring (i.e., Rho A, aniline, actin and myosin) (Harding et al., Reference Harding, Moccia, Drunat, Soukarieh, Tubeuf, Chitty, Verloes, Gressens, El Ghouzzi, Joriot and Di Cunto2016). This gene is located on chromosome 12q24.23, comprising 50 exons, 2027 amino acids, with a genome size and molecular weight of 191,501 bp and 231,431 Da, respectively. It has four already known isoforms and a 6207 bp ORF. The CIT protein is located in the mitotic cells midbody and cleavage furrow. This gene is conserved in many species including H. sapiens, M. musculus, D. melanogaster, M .mulatta, P. troglodytes, X. tropicalis, X. tropicalis and A. gambiae. CIT is important in phosphorylation of many compounds, for the maintenance of the structure of the midbody during cell division and in the completion of cytokinesis due to kinase activity (Di Cunto et al., Reference Di Cunto, Imarisio, Hirsch, Broccoli, Bulfone, Migheli, Atzori, Turco, Triolo, Dotto and Silengo2000; Li et al., Reference Li, Bielas, Zaki, Ismail, Farfara, Um, Rosti, Scott, Tu, Chi and Gabriel2016). Homozygous mutation in the kinase domain of CIT causes MCPH by the loss or activation of protein citron kinase (Basit et al., Reference Basit, Al-Harbi, Alhijji, Albalawi, Alharby, Eldardear and Samman2016; Harding et al., Reference Harding, Moccia, Drunat, Soukarieh, Tubeuf, Chitty, Verloes, Gressens, El Ghouzzi, Joriot and Di Cunto2016). Individuals with these mutations exhibit abnormal cytokinesis characterized by deferred mitosis, spindles having multiple poles, chromosomal instability, aneuploidy, PF3 cycle arrest initiation, massive apoptosis and have multinucleated neurons throughout the cerebral cortex (Li et al., Reference Li, Bielas, Zaki, Ismail, Farfara, Um, Rosti, Scott, Tu, Chi and Gabriel2016). CITK deficient cells have heightened levels of sensitivity to ionizing radiation and inoperative rehabilitation from radiation-induced DNA damage (Bianchi et al., Reference Bianchi, Tocco, Pallavicini, Liu, Vernì, Merigliano, Bonaccorsi, El-Assawy, Priano, Gai and Berto2017). Mutations in the CIT gene causes a severe neurological disorder characterized by very small head and individuals with this microcephaly have intellectually disability, axia hypotonia, brisk reflexes, social impairment, thin corpus callosum and reduced cerebral volume and sometimes have a sloping forehead, dysmorphic features and their brain shows gyral patterns (Harding et al., Reference Harding, Moccia, Drunat, Soukarieh, Tubeuf, Chitty, Verloes, Gressens, El Ghouzzi, Joriot and Di Cunto2016; Li et al., Reference Li, Bielas, Zaki, Ismail, Farfara, Um, Rosti, Scott, Tu, Chi and Gabriel2016; Shaheen et al., Reference Shaheen, Hashem, Abdel-Salam, Al-Fadhli, Ewida and Alkuraya2016). In rodents CITK is important for the proliferation of neural progenitor cells and male germ cell precursors. CIT knockout mice show ataxia, testicular hypoplasia, lethal seizures, growth deficiencies, severe reduction in brain size and often show simplified gyral patterns and temperaments linked to cytokinesis defects, that is, they have multinucleated neurons throughout the cerebrum and cortex and show massive apoptosis (Harding et al., Reference Harding, Moccia, Drunat, Soukarieh, Tubeuf, Chitty, Verloes, Gressens, El Ghouzzi, Joriot and Di Cunto2016; Li et al., Reference Li, Bielas, Zaki, Ismail, Farfara, Um, Rosti, Scott, Tu, Chi and Gabriel2016; Bianchi et al., Reference Bianchi, Tocco, Pallavicini, Liu, Vernì, Merigliano, Bonaccorsi, El-Assawy, Priano, Gai and Berto2017).
(xviii) MCPH18 (WDFY3)
WDFY3, encoding Alfy protein, performs binding and beta-N-acetylglucosaminylglycopeptide beta-1,4-galactosyltransferase activity. Alfy protein domains are: WD40_repeat domain, WD40/YVTN domain and FYVE type domain. This gene is located on chromosome 8p23, comprising 3526 amino acids, with a genome size and molecular weight of 296,855 bp and 395,258 Da, respectively, and two already known isoforms. WDFY3 is used as a scaffold protein required for the selective self-breakdown of macromolecules such as aggregation-prone proteins. Alfy protein is highly expressed in the developing CNS and crucial for the development of axonal tracts. The protein breakdown is mandatory to provide the proper atmosphere in which to coordinate complicated cell signalling events and to enhance cellular remodelling. The Wdfy3 gene is suspected to be involved in neurodevelopmental ataxia such as microcephaly and autism. Its homolog is Blue Cheese (bchs), which is abundantly expressed in the developing and adult fly CNS. Wdfy3 dysfunction in mice has subtle effects on migration and proliferation of neural progenitor cells. The affected mice display larger brains as a repercussion of an analogous alteration in the mechanism of radial glial mitoses to symmetric from asymmetric. The other 17 genes of microcephaly follow the autosomal recessive mode of inheritance; however, the WDFY3 gene follows the autosomal dominant mode of inheritance. Heterozygous mutation has been reported in the WDFY3 gene (R2637W), a c.7909C-T transition means tryptophan replaces arginine at the 2637 position. In vitro expression studies on Drosophila have revealed increased levels of DVL3 upon transfection of Drosophila cells with mutant human WDFY3. Increased levels of DVL3 cause abnormal activation of Wnt signalling as well as continued generation of apical progenitor cells without transition to differentiation and generation of the basal progenitor cell layers in the cerebral cortex, thus resulting in impaired cortical development and microcephaly (Jayaraman et al., Reference Jayaraman, Bae and Walsh2018).
3. Conclusion
Microcephaly is considered to be a rare neurodevelopmental disorder with many underlying causes. Pathogenicity analysis led to the discovery of 18 genes being a possible reason for this primary neurogenic mitotic syndrome. Recent experiments for successful generation of model animals for MCPH genes opened the door for researchers to further comprehend the pathophysiology and aetiology of MCPH. Improvement in the genotypic and imaging techniques along with organizing patients according to the basis of their genotypic homogeneity would enable us to better predict a correlation between genotype–phenotype. Mutational analysis of patients from different regions of the Middle East, and especially Pakistan, would aid in counselling and diagnostic approaches for MCPH ensuring better health quality for patients. This study has allowed us a better understanding of neuronal production processes by stem cells. The ongoing research projects on MCPH genes will lead us towards better understanding of this rare nonprogressive neuropediatric disorder. Moreover, MCPH genes are strong candidates for brain development and evolutionary studies.
Declaration of interest
None.