CDKL5 deficiency disorder (CDD) has been increasingly recognized in the last decade as a distinct clinical entity (OMIM #300672), presenting in early infancy with seizures, severe developmental delay, loss of milestones, and postnatal microcephaly.1 This disorder was originally described in the mid-2000s motivated in part by the heterogeneity of the loose grouping of diseases referred to as atypical Rett syndrome (RTT), variant RTT, or Rett-like syndromes.Reference Evans, Archer and Colley2
Several seizure types have been associated with CDD, particularly “hypermotor-tonic-spasms” sequence; however, water-related reflex seizures have not been associated with CDD. Reflex seizures are defined as seizures precipitated by external or internal stimuli.Reference Engel3 Hot-water epilepsy or water-immersion-induced seizures are unique types of reflex seizures and have not previously been reported in CDD. In this paper, we describe a novel presentation of a patient with confirmed CDD and water-immersion reflex seizures.
The patient is the first child of non-consanguineous healthy parents with a negative past medical history and family medical history. The pregnancy and birth history were also unremarkable and the child was born at term via viginal delivery. There were no neonatal complications. She first presented at 6 months of age with onset of seizures provoked by bathing (water immersion). She was being bathed by her mother when she turned her head to the right with a fixed stare and her whole body was hyperextended in a tonic posture followed by generalized tonic–clonic movements. The entire episode lasted about 30 sec, returning to normal afterwards. Subsequently, similar episodes occurred lasting 30–60 sec, and only during bathing (Video 1 in Supplementary Material). On physical examination, she was non-dysmorphic, alert but irritable. General examination was normal, particularly growth parameters which remained at 25–50th percentile throughout her growth. There was axial hypotonia and poor visual tracking. Axial hypotonia remained consistent until present. Touch sensation, peripheral tone, strength, and deep tendon reflexes were normal. She was started on phenobarbital with modest improvement. The frequency of seizures decreased after limiting full bathing using wet cloths instead. An electroencephalogram (EEG) performed at 8 months of age showed high-amplitude background activity, somewhat poorly developed sleep features and intermittent asymmetric parietal activity, indicating very mild diffuse brain dysfunction (Figure 1). Brain magnetic resonance imaging with spectroscopy was normal. Metabolic investigations in blood, urine and cerebrospinal fluid, and oligonucleotide array-comparative genomic hybridization (aCGH) were all normal. She sat independently at 8–9 months of age. Her tracking seemed inconsistent, improving 6 months later. At 12 months of age, rhythmic repetitive midline hand wringing movements were reported. Sequencing of MECP2 was normal. Targeted aCGH with exon-level resolution of CDKL5 was carried out (GeneDx, Gaithersburg, MD), which detected a deletion encompassing exons 2–3, which was confirmed by Sanger sequencing. At 16 months old, she developed 3–10 min long clusters of subtle epileptic spasms triggered by immersion or noises first but later were unprovoked (Video 2 in Supplementary Material). At 20 months of age, axial hypotonia had improved, but she became hyperreflexic in all limbs. She had social smile and language at a 3–4-month level with some non-specific vocalizations. At this time, 2–3 times per week her seizures evolved into generalized tonic–clonic with pronounced acrocyanosis (Video 3 in Supplementary Material). Acrocyanosis was also seen independently. Repeat EEGs at 22 and 52 months of age showed multiple independent spike foci and slow background activity for age, indicating epileptic encephalopathy (Figures 2 and 3, respectively). She required glasses by 36 months of age. Various combinations of topiramate, valproic acid, oxcarbazepine, and clobazam had failed to control her seizures. Ketogenic diet was declined. At 7 years old, she is able to stand with assistance, push herself to ambulate while strapped in a walker device and is non-verbal. She requires bilateral orthotics for Achilles tendon contractures and still has moderate axial hypotonia.
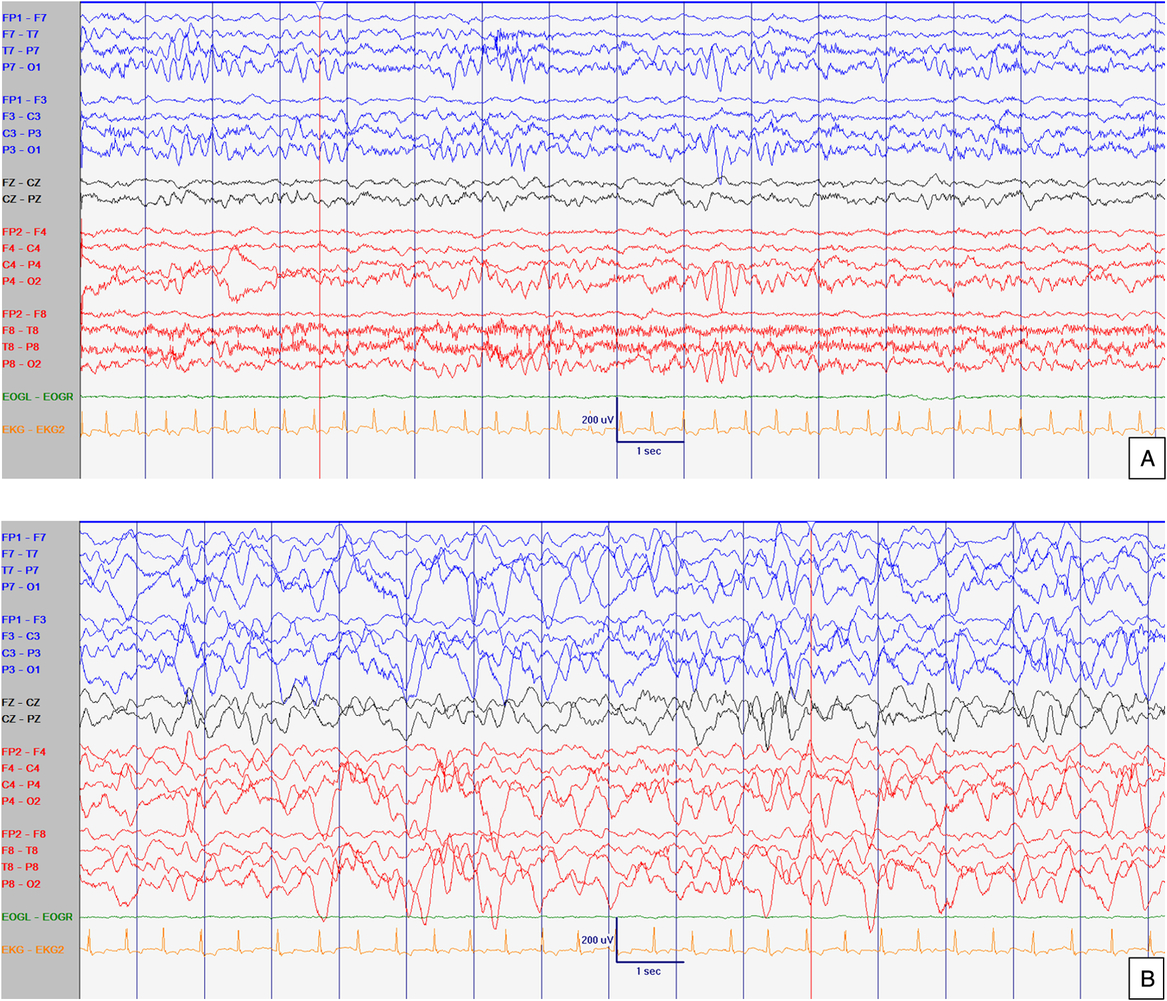
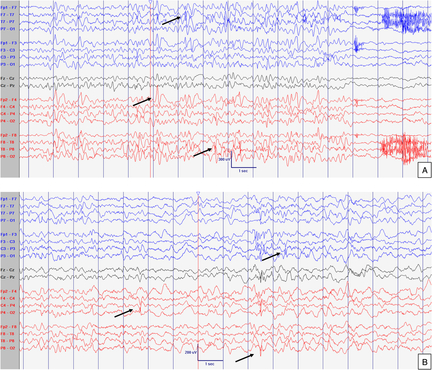
Figure 1: EEG at 8 months old. Normal interictal background activity (5 Hz PDF) in awake (A) and sleep (B) states. Sensitivity at 10 uV/mm. LFF 1Hz. HFF 70 Hz. Notch OFF. Time base 30 mm/sec.
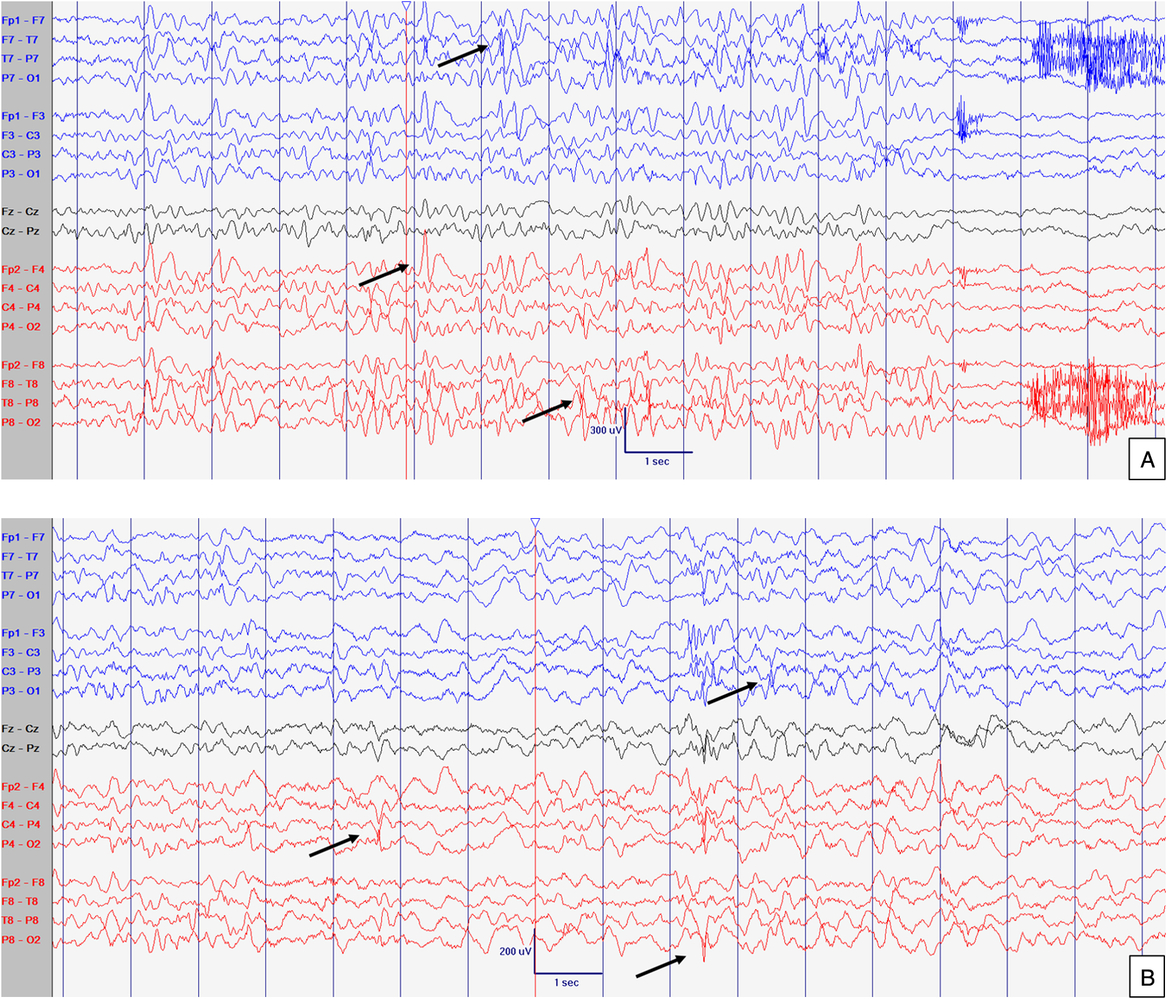
Figure 2: EEG at 21 months old. Abnormal interictal background activity (slow and dysregulated PDR for age) with occasional bilateral independent sharps and spikes with maximum negativity mainly over temporal areas but also frontal head regions (arrows) in awake (A) and sleep (B) states, particularly in R1. Events not captured. Sensitivity at 15 uV/mm. LFF 1Hz. HFF 70 Hz. Notch OFF. Time base 30 mm/sec.
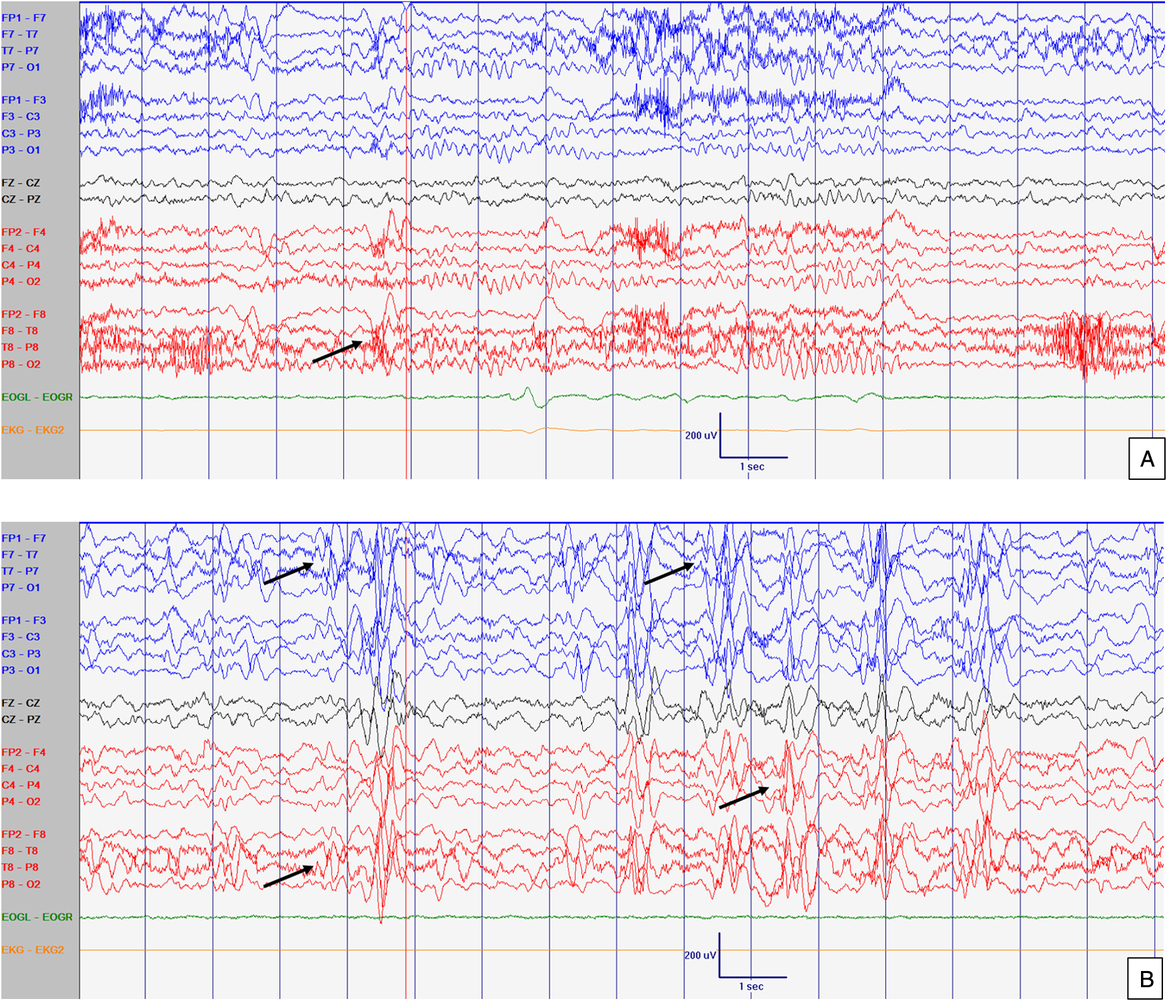
Figure 3: EEG at 52 months old. Abnormal slow background activity, but reactive to eye opening and closure during awake with slow sharply contoured (left > right) temporal activity (A) which increased in amplitude and in frequency during sleep (B). Arrows point abnormal interictal discharges. Sensitivity at 10 uV/mm. LFF 1Hz. HFF 70 Hz. Notch OFF. Time base 30 mm/sec.
CDD has been historically characterized by seizures and Rett-like features that emerge in the first year of life.Reference Neul, Kaufmann and Glaze4 Although RTT and CDD share many features, there are important differences, both from a clinical and pathophysiological standpoint. In CDD, one of the first features is seizure onset by 3–6 months of age, in contrast to RTT, which usually presents with developmental or behavioral features with seizures developing after 1 year of age if at all.Reference Neul, Kaufmann and Glaze4 The seizure semiology can be distinct in CDD as it evolves with age, including the unique hypermotor-tonic-spasm sequence that has been well described.Reference Bahi-Buisson, Kaminska and Boddaert5, Reference Klein, Yendle and Harvey6
Reflex seizures associated with CDD have not been previously reported in the literature and are rarely seen in children with RTT.Reference Martínez, Colmenero, Pereira, Vilaplana, Morón and Marfa7 In CDD and RTT, it can be very difficult to distinguish epileptic from non-epileptic events especially given the breathing abnormalities, dysautonomic features, and abnormal movements that can mimic seizures; therefore, it is possible that the prevalence of reflex seizures may be much higher. Interestingly, our patient presented with reflex seizures in infancy, whereas the three patients reported with RTT developed reflex seizures in adolescence, two to hand pressure (one with confirmed MeCP2 mutation) and one to eating.Reference Martínez, Colmenero, Pereira, Vilaplana, Morón and Marfa7 The discordance between the ages of onset may be related to the underlying pathophysiologies in both syndromes. Central nervous system hyperexcitability, thought to be associated with reflex seizures, may precede the diffuse epileptogenic potential that is responsible for the spontaneous seizures as it was seen in our patient. In contrast, the appearance of reflex seizures later in RTT after the plateau period may be explained by the delayed appearance of dysregulated multigene transcription, as seen in hypothalamic neurons, leading to neuronal network dysmaturation, autonomic dysfunction, and cortical hyperecitability.Reference Rudensky, Piccirillo and Mendez8, Reference Singh and Santosh9
Reflex seizures are described in other epileptic syndromes secondary to a known genetic disorder such as Down syndrome where nearly 20% of patients report reflex seizures.Reference Okudan and Özkara10 Its cortical hyperexcitability is thought to be secondary to similar findings to CDD (decreased density and size of synapses, dendritic spines of pyramidal neurons, lack of growth of dendritic trees, and reduction of neuronal density).Reference Okudan and Özkara10 Other syndromes with reported reflex seizures include, but do not limit to, Angelman syndrome, Juvenile Myoclonic Epilepsy, SCN1A, LGI1, FOXG1, and Synapsin 1 mutations.Reference Okudan and Özkara10, Reference Italiano, Striano and Russo11 They could be focal or generalized and appear similar to unprovoked seizures with the exception that a consistent trigger can be identified. The most common trigger factor is visual stimuli in up to 75% of the cases.Reference Okudan and Özkara10 The different reflex seizure types are ultimate expressions of excitatory and inhibitory dysregulation mechanisms in specific corticosubcortical and thalamocortical pathways with a common outcome: the activation of a critical neuronal mass that eventually results in a seizure when the seizure suppression threshold is exceeded.Reference Koepp, Caciagli and Pressler12
Treatment of CDD is symptomatic, and anti-seizure medication, vagal nerve stimulator, and the ketogenic diet are mainstays with variable success. It is very difficult to recommend specific agents to achieve seizure control given the complex and evolving nature of the epileptic encephalopathy and limited data of consistent long-term pharmacologic success. Regardless of etiology, trigger avoidance is recommended in the management of all reflex seizures, and it was partially effective in our patient. Nonetheless, this is not always feasible especially if the reflex seizures are provoked by internal stimuli.Reference Okudan and Özkara10
Herein, we describe a patient with CDD who initially presented with reflex seizures. This should aid the clinician to suspect CDD in young infants with seizures. Despite the possibility of achieving some control of reflex seizures in CDD and RTT, the prognosis for spontaneous seizure control remains poor. Therefore, in all infants who present with reflex seizures, the prognosis should be guarded until an underlying syndromic cause is identified as these seizures could also be seen in less ominous epileptic syndromes (photosensitive seizures in genetic generalized epilepsies).Reference Singh and Santosh9 Thus, this report could impact on diagnosis, treatment, and counselling in similar situations.
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/cjn.2019.29.
Acknowledgements
We thank the family, EEG staff and colleagues for allowing us to participate in the care of their child and their helpful discussions about the topic allowing us to learn together.
Statement of Authorship
TP collaborated with data collection, generated the first version of the manuscript and collaborated in subsequent revisions. JNH collaborated with data collection and manuscript revisions. AAM collaborated with manuscript revisions. CRG collaborated with manuscript revisions. JPA generated the first observation, guided TP throughout his work collaborating in data collection, manuscript generation and subsequent revisions.
Disclosures
CRG reports personal fees from Alexion Pharmaceuticals, outside the submitted work. The other authors have no conflicts of interest to disclose.