

Introduction
After more than 5 years in the making, a final revision of the federal regulations regarding human subjects research was released in January 2017 (https://www.gpo.gov/fdsys/pkg/FR-2017-01-19/pdf/2017-01058.pdf) [1] with a possible implementation target date in 2018. This Federal Rule for the Protection of Human Subjects, also referred to as the “Common Rule,” is the largest scale revision since 1991 [Reference Bierer, Barnes and Lynch2]. Many of the changes will require institutional review boards (IRBs) to reassess and revise their standard operating procedures, but one of the motivators for the Common Rule revisions is an attempt to facilitate and improve participants’ comprehension of protocols for which they are volunteering. Increasing research participant comprehension of study protocols is a laudable and important goal in light of considerable evidence that, despite numerous efforts and interventions, participants are often unclear about many aspects of the research for which they have given “informed consent” [Reference Cohn and Larson3–Reference Moreira10].
One approach to improving the consent process is the development of simplified templates which can be used to improve the health literacy and general readability of consent forms. We conducted a recent review of Web sites from 144 Association of American Medical Colleges (AAMC)-accredited institutions in the United States, 21 institutes in the National Institutes of Health (NIH), and the World Health Organization (WHO). Although 105 (72.9%) medical schools, 3 (14.2%) NIH institutes, and the WHO had at least one publicly available consent form template, there was considerable variation and lack of standardization in their format and content and little published evidence of their effectiveness [Reference Jackson and Larson11]. Like many other academic health centers, we also noted that the reading levels of our own approved consent forms were higher than recommended standards [Reference Foe and Larson12, Reference Larson, Foe and Lally13]. Given that enhanced consent forms have been shown to improve research participant’s understanding and the interest in improving health-related human subjects research, we were interested in how informed consent forms could be improved to address the needs of both participants and the research community [Reference Nishimura14].
With federal funding (National Center for Advancing Translational Sciences, National Institutes of Health UL1 TR000040), we developed and implemented several measures to improve the process and content of the research consenting process. One such effort, which has been incorporated into the training of consent administrators, was the development and testing of a training module and video [Reference Larson15]. A second activity was to develop a series of consent form templates using the principles of health literacy. The aim of this paper is to provide results from surveys we conducted to evaluate the extent to which the consent form templates were known, used, and considered to be useful by research staff.
Materials and Methods
Study Design
This was a descriptive pre-post survey conducted before and after development and dissemination of consent form templates.
Sample and Setting
The study was conducted in an academic health center which has 6 IRBs—1 for genetic protocols, 1 for oncology, 1 for protocols that are eligible for expedited review, and the other 3 which review all other protocols. Protocols which meet the Office for Human Research Protections (OHRP) criteria for expedited review (https://www.hhs.gov/ohrp/regulations-and-policy/guidance/categories-of-research-expedited-review-procedure-1998/) are reviewed by 1 of 5 IRB chairs. Collectively, the IRBs process ~5000 active protocols, and the office is staffed by ~22 fulltime human subject research staff members.
Template Development and Testing
Templates were developed and trialed by a group of experts in clinical research, health literacy, national regulatory requirements, and end users. The template development team’s initial task was to outline the process for decision making and testing of the templates, agree upon roles and responsibilities of team members, and develop a timeline. The team then met monthly to review materials and reach consensus at each step. We chose to focus first on templates for expeditable protocols and assess them before developing templates for protocols requiring full board review.
First, we reviewed regulatory requirements regarding information to be included in consent forms. Based on this review, we determined that the templates would include 10 sections which reflected these mandated elements. We next decided to word these sections as questions that reflected what participants needed to know in an easily readable, accessible manner. These sections for the adult templates were: What information is on this form? Why is this study being done? What will I be asked to do if I choose to be in this study? Are there any risks? Are there any benefits? What about my privacy? Will I get paid or be given anything to take part in this study? Will I incur costs if I take part in this study? What are my rights if I take part in this study? The assent form for 9–11 year olds had the following headings: Why are we interested in talking to you? What will happen if you are in the study? Will it hurt? What if you have questions? Do you have to be in this study? The assent form template for older teens mirrored that of the adult form.
The initial drafting of the templates was done by one team member (Meyer) who is bilingual (English/Spanish), a recognized expert and consultant in health literacy, vulnerable populations, and cultural competence, and has mentored and taught health sciences students and trainees in these areas for several decades. Using her extensive community networks, she obtained feedback iteratively from community members, many of whom were first generation immigrants, non-native English speakers, and/or who had limited education (high school or less), regarding the readability and clarity of the forms. Edits and changes were made as suggested to assure that text was understandable and consistent in meaning. Drafts were reviewed by the entire study team as they were developed. Next, a team manager in the institution’s Office of Human Research Protection reviewed and edited the templates to assure that regulatory and local requirements were included. Over a period of about 2 years (2012–2014), 4 templates written at the eighth-tenth grade reading level were completed: 1 for minimal risk research, a second for minimal risk research that included audio or video recording, and 2 assent forms for children 9–11 and 12–17 years of age.
Template Implementation
After their development and feasibility testing, the templates were added to the IRB Web site and IRB staff were made aware of their availability, but there was no active, formal dissemination of information about the templates to researchers. After the first year in which templates were “passively” available, additional dissemination efforts included discussion of templates at investigator meetings, placement of templates in a more accessible location on the IRB submission pages, and suggestions to consider using templates from IRB staff to researchers during their pre-review process.
Data Collection
We conducted email and telephone surveys of researchers at 2 time points: in January-March 2015 after the templates had been available on the IRB Web site for about 6 months without any formal dissemination efforts and again in February-March 2017 following the additional dissemination efforts described above. Following approval from the Columbia University Medical Center IRB, we obtained a list of the most recent new protocols (n=124 in baseline survey and 100 in follow-up survey) which had been approved under expedited review procedures by any of the 6 IRBs and included a consent form. Protocols that received a waiver of written documentation of consent were excluded.
To determine who had actually used a template, emails were initially sent from the IRB office to the submitter of the protocols, which was generally either the principal faculty investigator, a graduate student investigator, or a research staff member. After 2 weeks, nonrespondents were contacted by telephone by one of the authors to maximize response rate. Second, for those who responded that they had used a template, a follow-up user’s evaluation survey was conducted to determine how the forms were accessed, whether they were easy to find and facilitated the IRB review process, and to solicit recommendations for improvement.
Survey Questions
The initial emailed survey described the purpose of the study and the templates and included 2 questions: Have you heard of these templates? If yes, have you used them? If no, do you know where you might find them? Those who had used templates were asked to provide feedback on their usefulness and ease of use.
Analysis
Results were summarized descriptively and χ2 tests were used to compare responses between the initial and follow-up surveys.
Results
In the initial survey (2015), 86/124 (69.4%) of surveyed researchers responded, 18 (20.9%) of whom had heard of the templates and 5 (5.8%) reported that they had used them. In the follow-up survey 2 years later, the response rate was 59.0% (59/100); 54.2% (32/59) had heard of the templates and 87.5% (28/32) had used them. Rates of respondents who had heard of or used templates were significantly higher in the follow-up survey (both p<0.0001), see Table 1.
Table 1 Responses regarding use of consent form templates at baseline (January-March 2015) or follow-up (February-March 2017)
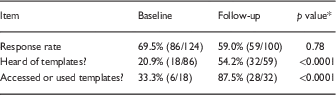
* χ2.
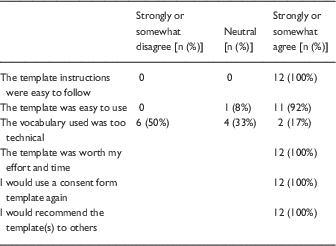
In the follow-up survey, half of respondents (14/28) who reported that they had used the templates and were sent a follow-up survey provided their feedback. The majority of respondents (11/14, 79%) were physicians; the others were from nursing or public health. The majority (10/14, 71.4%) were faculty or research administrators and 28% (n=4) were students/trainees who had learned about the templates from the web (71%) or from peers (29%). In total, 12 researchers who had used the templates completed a satisfaction survey, and all agreed that the templates were useful, that they would use them again and recommend them to others. On the other hand, half of respondents still reported that the template vocabulary was too technical for participants (Table 2). One researcher reported that his protocol was returned by the IRB before approval because the original consent form did not include the required elements. After using the template as requested, the form was approved.
Table 2 Respondent reactions to use of templates (n=12)
Discussion
Template Development
The development and testing of the templates was longer and more arduous than we anticipated, requiring several years of work to assure that they met basic health literacy criteria (https://www.cdc.gov/healthliteracy/learn/), were valid and reliable, acceptable and understandable to research participants, vetted and approved by the IRB, and readily accessible to investigators. We encountered unexpected challenges primarily at 2 points. First, the staff of the institution’s Human Subjects Research Protection Office were exceedingly busy and the team manager designated to do a final assessment of the templates for regulatory compliance and consistency with local requirements needed several months to complete a final assessment and have the templates vetted by administration. Second, the technical requirements to incorporate the templates into the electronic protocol submission process also required considerable time and staff resources. Institutions considering the adoption/adaption of consent form templates need to be cognizant of such resource, political and technical issues and assess the cost-benefit and “value-added” of templates, particularly those developed in-house.
Template Implementation
As these templates were “rolled out” and made available to researchers, it was important to assess their use at baseline and after a period of time to allow for their dissemination. Our initial “dissemination plan” was passive; we simply added the templates to the IRB electronic protocol submission form. It was clear after the baseline period that a more active marketing approach to bring the templates to the direct attention of researchers was necessary. The results after a year of more intensive marketing were positive, with templates being used significantly more often in the follow-up period and researchers reporting that they were useful and efficient. Clearly, adopting any change takes time and resources, and the introduction of new forms or processes often requires considerable effort. Some researchers also reported that they did not use the templates because they had their own templates from previous studies.
There were limitations to this study. As with any self-report data, it is possible that nonresponse or social desirability biases skewed the findings. It seems likely that these biases, however, would be nondifferential as they would have affected both the baseline and follow-up surveys equally. Because we did not include information about the investigators who responded to the surveys, it is possible that some individuals who responded to the first survey were also included in the second. If that were the case, it could introduce bias into the response regarding whether they had heard of the templates. We decided, however, that it was more important to reduce social desirability bias by assuring researchers that their responses would not be linked to them.
Finally, despite this major effort, some researchers still judged the language to be too technical for participants. We did not obtain information regarding the types of patients for whom these templates were used, nor did we review the consent form sections populated by research staff to assess the reading level of those added sections; this content may have contributed to the vocabulary used being too technical for participants to understand. Whether templates would be more effective if they were population-specific beyond age and primary language may be an area for future research.
Lessons Learned and Recommendations
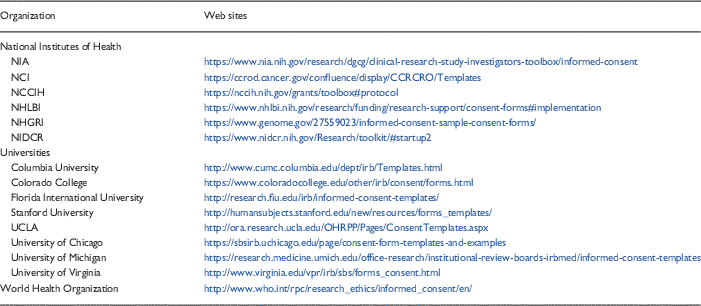
Templates are one promising tool that may improve the consenting process. As noted above, however, the implementation and dissemination of the templates (e.g., getting them uploaded onto the IRB Web site in a convenient and easy to find location and marketing their availability to research staff) required months of concerted effort. The decision to adopt or adapt templates therefore must include an understanding of what is required to assure their sustainability within the system. Many templates are already available from a variety of sources—NIH, medical school or university Web sites, the WHO for example (see Table 3 for samples). Hence, it seems reasonable to first consider using available resources rather than attempting to develop new templates “from scratch.”
We recommend the use of consent form templates as 1 component of a broader, multi-faceted human subjects research protection program to improve efficiency for researchers and clarity and comprehension among research participants. Nevertheless, although a prior meta-analysis of controlled trials testing difference approaches to consent reported that some consent templates can improve comprehension of informed consent [Reference Nishimura14], the use of a template per se does not necessarily improve the patient’s understanding. The next step would be to assess the impact of templates on the actual effectiveness of consent from the perspective of the participant. Even for individuals who have previously participated in research, if they see the same “looking” forms in the same format over and over again, they may create a mental heuristic and simply agree without reading. So, although consent form templates may be helpful and easier to read, more research that steps outside the box to test other novel consenting processes is indicated. In the meantime, adopting templates already publically available and adapting them as appropriate for local use seems a prudent and efficient course. This is consistent with the intent of the revised Common Rule to facilitate and improve transparency in the informed consent process.
Acknowledgments
Partial funding for the initial development of the templates was provided by the National Center for Advancing Translational Sciences, National Institutes of Health UL1 TR000040 (D.M. and E.L.L.). The authors thank the staff of the Human Research Protection Office at Columbia University Medical Center and the researchers who responded to the surveys.
Disclosures
The authors have no conflicts of interest to declare.



 Open access
Open access