


FOCUS POINTS
• Alzheimer's disease (AD) is becoming more common as the world population ages.
• New treatments for AD are urgently needed.
• Translational neuroscience comprises the development of new treatments and diagnostic devices that will assist in diagnosing, preventing or treating diseases of the nervous system.
• Animal models of AD demonstrate efficacy and safety in pre-clinical settings and function as screens for agents to be advanced to human testing.
• Clinical trial programs include Phase 1 testing to establish human pharmacokinetics, Phase 2 assessments to demonstrate proof of concept and dose and Phase 3 trials to confirm efficacy.
• Biomarkers demonstrate the biological effects of disease modifying drugs and assist in AD drug development programs.
• Funding of translational research is changing with an increased emphasis on discovery in academic medical centers with support from philanthropy and advocacy groups leading to later stage in licensing by pharmaceutical companies.
• U.S. federal resources available to support AD drug development include the National Center for Advancing Translational Science (NCATS), the Clinical and Translational Science Award (CTSA) programs and the Alzheimer's Disease Cooperative Study (ADCS) among others..
Introduction
Alzheimer's disease (AD) is a neurodegenerative disease that progresses from mild cognitive impairment to severe dementia and death. AD is increasingly common with age, doubling in frequency every five years after the age of 60.Reference Qiu, Kivipelto and von Strauss1 AD is rapidly becoming a major challenge to public health, as well as a common personal catastrophe for patients and families as the world's population ages. Therapies that prevent or delay the onset, slow the progression, or improve the symptoms of AD are urgently needed. Five drugs are approved for the treatment of AD—tacrine, donepezil, rivastigmine, galantamine, and memantine—but no agents have been approved since 2004 despite many phase 3 trials.Reference Aisen, Cummings and Schneider2 There is increasing concern about the difficulty of developing drugs with disease-modifying potential and with the high costs associated with AD drug development.Reference Naylor, Karlawish and Arnold3, Reference Iliffe4
The challenges associated with AD drug development occur in the context of shifts that are occurring in how science is organized, with an emphasis on translational research and translational medicine. Translational research is usually divided into four stages (T1–T4) that link basic science to clinical science and clinical science to the practice of medicine and public health outcomes, respectively.Reference Woolf5 T1 addresses the transfer of knowledge of disease mechanisms into the development of new methods for diagnosis, treatment, or prevention of disease; T2 refers to the translation of results of clinical studies into clinical practice and decision making.Reference Woolf5 T3 addresses diffusion and implementation in community practice, and T4 assesses real-world outcomes on public health.Reference Kahn and Weng6 Translational research focuses on the development of new devices, drugs, and diagnostics that will have benefit to people in the short or long term. Translational neuroscience comprises the development of new treatments and diagnostic devices that will assist in diagnosing, preventing, or treating diseases of the nervous system. Much of AD drug development is embraced by the concept of T1 translational neuroscience. Translational neuroscience includes animal models of AD, biomarkers for AD, and clinical trials for AD diagnostics and therapeutics. Translational neuroscience, when successful, leads to the development of products (drugs, devices), and there is a commercial application for this aspect of science. In this article, we address the challenges of AD drug development, how translational neuroscience approaches may be applied to AD drug development, and how financial and commercial aspects of AD drug development are integrated into the decision framework.
Alzheimer's Disease Drug Development
The first successful drug development program for AD culminated in the approval of tacrine, a cholinesterase inhibitor. Other cholinesterase inhibitors with improved safety profiles or formulations followed, with the approval of donepezil, galantamine, and rivastigmine and rivastigmine patch. Recently, high-dose options for donepezil and rivastigmine have been approved.Reference Farlow, Veloso and Moline7, Reference Cummings, Froelich and Black8 Memantine is an N-methyl-D-aspartate (NMDA) antagonist that was approved for the treatment of AD in 2004. No other classes of agents have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of AD.
There have been many failures in AD drug development (Table 1). In some cases, the absence of a drug–placebo difference at the trial's end reflected failures of the trial as suggested by the absence of decline in the placebo group, excessive measurement variability, or failure to demonstrate a treatment effect in an active comparator arm of the study using donepezil.Reference Cummings9 In other trials, the failure of the program to lead to an approvable agent could be ascribed to lack of efficacy or safety.
Table 1 Agents that completed phase 3 trials for AD and showed no drug-placebo difference on prespecified primary outcomes

Translational Neuroscience: Model and Key Concepts
Figure 1 shows the steps of drug development. The process begins and ends with human disease. Patients are identified as suffering from a disease, and study of the disease leads to targets that are possibly amenable to therapeutic manipulation to prevent or slow the disease process. Candidate therapies are identified in assays, typically by high throughput screening in which thousands to millions of compounds are screened in an assay to identify “hits” that may be developed into “leads” that may eventually be optimized into candidate therapies. These basic science steps are not necessarily included in the concept of translational research, although the quest to find assays that better predict human efficacy and toxicity blurs the boundaries between basic and early translational science. For example, in silico structure–activity relationship (SAR) modeling and dynamic molecular simulations are used to identify compounds for AD that are most likely to inhibit amyloid-beta aggregation and related neurotoxicity.Reference Connelly, Jang and Arce10 SAR modeling of c-Jun-N-terminal kinase (JNK) inhibitors has aided in identification of brain-penetrant compounds that are predicted to be more effective in preventing neurodegeneration.Reference Bowers, Truong and Jeffrey Neitz11 High-content screening in intact cell lines is increasingly used to identify promising compounds, and “humanizing” this approach through the use of induced pluripotent stem cells (iPS cells) builds a physiologic bridge between the screen and human application.

Figure 1 Overview of translational research as a framework of drug development.
Once a lead compound or group of related compounds is identified that has promise in the screening assays and has properties that support “druggability”—acceptable physical and chemical properties that suggest they might be developed as therapies—then pharmacokinetic and pharmacodynamic studies of the agents can be initiated. Pharmacokinetic studies establish the absorption, distribution, metabolism, and excretion (ADME) of the agent, and toxicity studies search for cardiac, pulmonary, liver, endocrine, renal, skin, muscular, and nervous system effects that might disqualify the agent for further study. Development of neurotherapeutic compounds in the translational neuroscience paradigm has the additional challenge posed by the need to penetrate the blood–brain barrier. Long-term carcinogenic studies and research that might indicate reproductive toxicity and teratogenicity are pursued. At least two species are assessed for most potential effects, and species known to be particularly sensitive to some adverse events (e.g., dogs in the case of cardiac effects) are employed. Again, these pharmacologic studies are not necessarily of a translational nature, but to the extent that they can be made to be more predictive of human ADME and toxicity, the more translational value this research assumes. Recent progress in predicting renal toxicity using biomarkers is an example of translational research that advances early-stage drug development.Reference Marrer and Dieterle12
Preclinical pharmacodynamic assessment typically involves determining the effect of the candidate agent on an animal model of the disease. For AD, aged animals, senescence accelerated animals, chemical- and lesion-induced rodent models, and transgenic species (mice, rats, fruit flies, and others) comprise the animals in which testing occurs.Reference Van Dam and De Deyn13 Combinations of animals and sequential testing in model systems may be more predictive of human efficacy and provide more insight into the range of effects of the agent,Reference Sabbagh, Kinney and Cummings14 but no animal model recapitulates all aspects of AD, and none have yet successfully anticipated a beneficial effect in subjects with AD. Readouts of the effect of the test agent include behavior (i.e., Morris Water Maze, fear conditioning, novel object recognition), histology (i.e., number of plaques), and biochemistry (i.e., total amount of amyloid beta-protein [Aß]). Dose–response relationships are explored. Improving the predictive value of animal models is a key component of translational neuroscience as applied to AD drug development. There are many aspects to this translational challenge: species-to-species relationships, timing of intervention, dose equivalency, pathways affected, duration of treatment, genetic background, genetic contribution to the pathophysiology (e.g, transgenic species are humanized with known human mutations, but most patients with AD do not have a disease-causing mutation). Biomarkers applied in animals and then advanced to humans may provide necessary bridges between preclinical and clinical observations that have not yet been fully exploited. Animals models are a key element of translational research.
Once safety and efficacy have been established to an acceptable level in preclinical studies, the compound is advanced to human testing. First-in-human phase 1 studies involve testing single and multiple ascending doses beginning with doses typically 10-fold lower than the no observable adverse event level (NOAEL) in animals, adjusted to human doses by allometric scaling from the most sensitive species assessed. Human pharmacokinetics of the test agent are established in phase 1, and the maximum tolerated dose is also determined. Approximately, 50% of compounds are terminated at this point in development.Reference Paul, Mytelka and Dunwiddie15
Phase 2 clinical trials establish proof of concept (phase 2a) and the dose(s) (phase 2b) to be advanced to phase 3 trials. Phase 2 trials also expand the safety information available on the test agent. It is at phase 2a that there is the greatest influence of translational research. AD progresses slowly, and the sponsor is faced with the conundrum of doing a large lengthy study to establish clinical benefit or doing a shorter smaller study with a biomarker as the key outcome.Reference Cummings16 Biomarkers have smaller standard deviations of measurement and require smaller numbers to show drug-placebo differences.Reference van Rossum, Vos, Handels and Visser17, Reference Grill, Di and Lu18 There is substantial risk associated with this decision, since no biomarker has been proven to predict the clinical outcomes in AD trials. This is a central challenge for translational research because biomarkers are a focus of translational investigations. Biomarkers may have diagnostic value and become commercialized products of translational research (e.g., amyloid imaging with florbetapir; Figure 2), or they may be indicators of a therapeutic response used in drug development but not independently commercialized. In some cases, drugs and biomarkers are codeveloped in theranostic programs.Reference Blair and Blakemore19
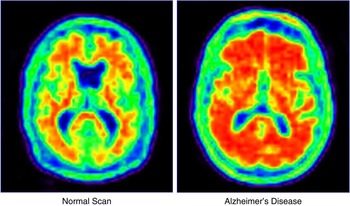
Figure 2 Florbetapir amyloid imaging in a healthy elderly person and an age-matched individual with Alzheimer's disease.
Approximately 35% of candidate drugs are progressed from phase 2 to phase 3.Reference Paul, Mytelka and Dunwiddie15 There is substantial controversy about how to define a phase 2 success. As noted, an effect on a biomarker may not predict a clinical response. Phase 2 outcomes that did not meet their primary endpoints may be interpreted as successful if a responsive subgroup is identified, but this strategy often results in failure to reproduce the subgroup findings in a larger trial.Reference Green, Schneider and Amato20 Well-conducted phase 2 studies will facilitate better understanding of the biology and pharmacology of the candidate, and will improve phase 3 success rates by allowing those agents with promise to be advanced and those with less promise to be stopped. Extending phase 2 to better understand the pharmacology and biology of the agent will result in more phase 3 successes. Improved trial designs and outcomes assessments are an important aspect of translational research to optimize the opportunity to develop successful treatments for AD.
Phase 3 confirms the observations of phase 2 in a larger number of patients and, if successful, leads to marketing approval of the new agent. Seventy percent of agents with positive phase 3 trials are prepared for FDA review; the overall chance of an agent entering phase 1 to be shown safe and effective and advanced to FDA review is 11%, and the attrition rate is higher for central nervous system drugs than for drugs in other therapeutic areas.Reference Paul, Mytelka and Dunwiddie15, Reference Talevi, Bellera and Di Ianni21 Once a new agent is available for widespread use, the later phases of translational research are inaugurated. T3 research refers to the translation of research into clinical practice, and T4 refers to the impact of the new intervention on public health.Reference Khoury, Gwinn and Ioannidis22 Evidence-based medicine refers to the practice of medicine as informed by double-blind placebo controlled trials and other data-driven methodologies.Reference Whitcomb23
Biomarkers in AD Drug Development
There are five primary types of biomarkers relevant to AD drug development: (1) brain imaging; (2) electrophysiologic measures; (3) plasma and cerebrospinal fluid (CSF) measures of prespecified analytes; (4) “omics” platforms with microarray and spectroscopic determination of multiple gene, protein, lipid, metabolite, or other measures combined with advanced informatics required to interpret the study results; and (5) genetics (Table 2).
Table 2 Biomarkers relevant to AD drug development

Brain imaging plays an increasingly important role in AD drug development (Table 3).Reference Cummings24 Magnetic resonance imaging (MRI) can be used to define a trial population, assess disease modification, or follow specific types of adverse events. MRI allows structural measures of the whole brain, ventricular system, or hippocampus; investigation of functional circuit activity with functional MRI (fMRI); measurement of white matter integrity with diffusion tensor imaging (DTI); assessment of blood flow with arterial spin labeling (ASL); and interrogation of neurochemical constituents with MR spectroscopy (MRS).Reference Li and Wahlund25 Positron emission tomography can be used with a variety of tracers: fluorodeoxyglucose (FDG) assessing, cerebral metabolism; fibrillar amyloid demonstrating the presence of neuritic plaques; aggregated protein to establish the presence of tau and amyloid (2-(1-{6-[(2-fluorine 18-labeled fluoroethyl)methylamino]-2-napthyl}ethylidene) malonitrile [FDDNP]); 5-HT1A receptors to show receptor function and neuronal integrity; verapamil measurement of p-glycoprotein function in the blood–brain barrier; oxygen measures of oxygen extraction; and translocation protein (TSPO; also known as the peripheral benzodiazepine receptor) assessments of microglial activation.Reference Clark, Schneider and Bedell26–Reference Van Assema, Lubberink and Bauer31 Single-photon emission computed tomography (SPECT) offers a measure of cerebral blood flow,Reference Kume, Hanyu and Sato32 as well as emerging measures of amyloid; dopamine transporter imaging can be used to exclude patients with dementia with Lewy bodies who have an AD-type phenotype.Reference Cummings33, Reference Cummings, Henchcliffe and Schaier34 MRI has played a critical role in detecting amyloid-related imaging abnormalities (ARIA) of the effusion and hemorrhagic type observed in the course of amyloid lowering clinical trials,Reference Sperling, Jack and Black35, Reference Sperling, Salloway and Brooks36 and is required as a safety measure in anti-amyloid treatment trials. Sample sizes required to show a drug–placebo difference with imaging are much smaller than those required to show clinical differences. For example, in a 6-month trial of an agent demonstrating a 20% drug–placebo difference in change from baseline, 257 patients would be required per arm to show the difference with ventricular atrophy, whereas 1370 would be required if the Alzheimer's Disease Assessment Scale—cognitive portion (ADAS-cog) was the outcome (these figures become 468 and 2100 for ApoE e4 noncarriers).Reference Nestor, Rupsingh and Borrie37 Neuroimaging is a critically important tool in translational neuroscience research for AD drug development.
Table 3 Neuroimaging in AD drug development

ARIA – amyloid related imaging abnormalities; ARIA-E – effusion; ARIA-H – microhemorrhage; FDDNP – 2-(1-{6-[(2-fluorine 18-labeled fluoroethyl)methylamino]-2-napthyl}ethylidene) malonitrile; FDG – fluorodeoxyglucose; MRI – magnetic resonance imaging; MRS – magnetic resonance spectroscopy; PET – positron emission tomography; SPECT – single photon emission computed tomography.
Specific CSF analytes have been extensively studied in AD, including Aβ-42, total tau, and phospho-tau (p-tau). The ratio of decreased CSF Aβ-42 to elevated tau or p-tau has high sensitivity and specificity for the diagnosis of AD.Reference Parnetti, Chiasserini and Eusebi38 Production of amyloid protein can be assessed with stable isotope labeled kinetics (SILK).Reference Bateman, Munsell and Morris39 Gamma secretase inhibitors have been shown to decrease Aβ production.Reference Bateman, Siemers and Mawuenyega40 SILK presents the opportunity to test target engagement and proof of pharmacology to help decide whether to advance agents in the drug development program. This is a direct translational neuroscience role. Analytes in blood have thus far been less useful in diagnosis and drug development of AD; however, studies of the potential translational application of inflammatory, oxidative, and other serum and plasma markers are being pursued.Reference Reale, Kamal and Velluto41, Reference Torres, Quaglio and de Souza42
Genetic studies focus on individual genes, whereas genomics refers to the entire genome or DNA sequence of organisms. Apolipoprotein E e4 is the gene variant that has the greatest impact on late-onset AD, increasing the risk of the disease and decreasing the age at onset.Reference Corder, Saunders and Strittmatter43 Other genes accounting for smaller percentages of the variance of AD risk but making identifiable risk contributions include CLU, PICALM, BIN1, SORL1, and CR-1.Reference Masoodi, Shammari, Al-Muammar and Alhamdan44, Reference Rogaeva, Meng and Lee45 Some of the risk genes identified have a role in amyloid beta-protein metabolism, but genetic observations also implicate immune system function, cholesterol metabolism, and synaptic membrane processes.Reference Morgan46 These observations point toward new avenues of drug discovery and development.
“Omics” strategies are another important component of contemporary translational neuroscience. RNA transcription of DNA leads to transcriptomics, which includes not only a comprehensive survey of the messenger RNA, but also noncoding RNAs such as micro-RNAs, which are emerging as key regulators of gene expression in normal and disease states. Proteomics refers to the analysis of proteins as they exist in the cell, revealing aspects of protein processing and post-translational modification that cannot be inferred from the corresponding DNA sequences alone. Metabonomics (metabolomics) involves the characterization of small molecules in circulatory or cell/tissue systems, and interactomics refers to the interactions among these levels.Reference Kandpal, Saviola and Felton47, Reference Morel, Holland and van der Greef48 All these omic signatures are based on expression arrays and mass spectrometric techniques that produce profiles of up-regulated and down-regulated expression of RNA, proteins, and metabolites found in tissues and fluids (blood, saliva, CSF).Reference Morel, Holland and van der Greef48 The metabolome includes the metabolites of molecules such as fatty acids, amino acids, nucleosides, steroids, and vitamins.Reference Morel, Holland and van der Greef48 Proteomic studies in AD suggest prominent involvement of inflammatory cell systems that may be relevant to treatment.Reference Hye, Lynham and Thambisetty49 Thus far, omic approaches have not informed drug development for AD, but these approaches promise to contribute to target identification and validation, predictive understanding of biological systems, monitoring of therapeutic responses, and eventual biological engineering.
The next step in utilizing omic data is the integrated analysis of genetic, genomic, protein, metabolite, cellular, and pathway event data into systems biology.Reference Morel, Holland and van der Greef48 The use of advanced mathematical strategies including informatics, biostatistics, data integration, computational biology, simulation and modeling, network analysis, and knowledge assembly is required to interpret the huge inventories of data generated by microarray and mass spectrometry studies.Reference Morel, Holland and van der Greef48, Reference Kholodenko, Yaffe and Kolch50, Reference Fischer51 This level of analysis is sometimes called quantitative biology, and systems and quantitative biology promise to become informative tools for drug discovery and development.Reference Fischer51 Translational informatics attempts to directly derive clinically relevant information from the vast omic data.Reference Tanaka52
Closely aligned with the concepts of omics and biomarkers is personalized medicine (also called precision medicine). In this approach, the unique biology of the individual patient is characterized in an effort to choose the right drug, in the right dose, for the right patient, given at the right time.Reference Tanaka52 Pharmacogenetics and pharmacogenomics are examples of precision medicine. Second-generation omics-based medicine will be predictive and is based on a thorough grasp of the complex manifestations of the disease from which the individual suffers.Reference Tanaka52 Omics-based medicine is increasingly providing a platform for translating quantitative systems biology into evidence-based medicine.
Biomarkers have several important roles in drug development (Figure 3).Reference Cummings24 They are used in drug discovery and in vitro assays to detect the effect of compounds in preliminary screens. They have important roles in preclinical drug development to detect evidence of efficacy or toxicity in animal models of the target disease. In preliminary human studies, they provide evidence of proof-of-pharmacology (POP) and target engagement (e.g., the SILK technique described above). In later stage clinical trials, they provide evidence of disease modification and proof-of-concept (POC) related to the putative mechanism of action (MOA) of the test agent. It is the hope of personalized (precision) medicine that biomarkers will eventually assist in choosing a responder and guiding dose, duration, and possible evolution of therapies over the course of the disease.

Figure 3 Role of biomarkers in drug development.
Funding Landscape for Translational Neuroscience and AD Drug Development
Translational research emphasizes products in the form of new diagnostic tests, drug treatments, devices, or processes that improve patient health through prevention or treatment. Products become available to broad populations through the processes of regulatory approval and marketing. The pathways by which the results of translational research become marketed products is being reshaped. In the traditional model, the National Institutes of Health (NIH) financed the basic science stages of idea development in academic settings, and then the product was either licensed to a company through university technology transfer offices or the inventor spun off a small biotechnology company and tried to raise capital to advance the product through angel funding (typically supporting very early development) and venture capital. If the product continued to show promise, the biotechnology company continued to seek venture capital or eventually had an initial public offering (IPO) and became publically owned with an infusion of capital sufficient to continue to progress the product toward market. Alternatively, a company with a promising product or the biotechnology company might be purchased by a major pharmaceutical company as a means of supplying the internal product pipeline of the pharmaceutical company. The difficulty of raising funds for the late preclinical and early clinical phases of development (ADME, toxicity, early stage human trials) gave rise to the name of “valley of death” for this stage of product development.Reference Beach53–Reference Finkbeiner55 In this model, angel funding, venture capital, biotechnology companies, and pharmaceutical companies all played critical roles in the financial ecosystem for drug and device development.
The low rate of success of drug development—especially CNS drug development—has led to marked changes in the approach to funding and more changes are anticipated. It is more difficult to attract venture capital to biotechnology endeavors, and pharmaceutical companies desire more advanced compounds and more well developed data packages before in-licensing, partnering, or purchasing a product.Reference Havenaar and Hiscocks56 De-risking compounds through the stage of POC or even phase 2 data is required by most pharmaceutical companies before they consider acquiring a candidate drug.
Alternative financial models are emerging. Pharmaceutical companies are working more closely with academic researchers and funding research in exchange for the right to develop products of interest.Reference Silber57 This increases opportunities for academic researchers to increase their involvement in early-stage translational research, but it imposes new demands in terms of reproducibility, scalabilty, intellectual property, and conflict of interest that impact the research. Some pharmaceutical companies are also funding nonprofit research institutes—such as Calibr funded by Merck in California—that will pursue research intended eventually to feed product pipelines. States and national governments are also starting or supporting venture capital firms intended specifically to support biotech and to fill the void left by traditional capital sources.Reference Reeve58 New approaches to funding are emerging, such as supporting one specific part of the drug development cycle.59
Philanthropy can play a critical role in drug development by supporting programs that can advance a drug or product from one stage to another. Philanthropic funding tends to be limited compared to industry, venture, and federal sources. Venture philanthropies “invest” in projects and share in intellectual property ownership and licensing or milestone payments if the compound is licensed, partnered, or sold.Reference Hanson, Nadig and Altevogt60 The Alzheimer Drug Discovery Foundation (ADDF) has promoted this funding model for AD drug discovery and development.Reference Lane, Shineman and Fillit61, Reference Refolo and Fillit62
Advocacy groups are also taking a greater role in drug development. This is particularly evident among advocacy groups for rare diseases, but AD groups also assist in drug development through programs such as the Alzheimer's Association's trial-match program and policy advocacy undertaken by the Alzheimer's Association, Alzheimer's Foundation of America, USAgainstAlzheimer's, and others.
Large clinical systems can capitalize on their high patient volume, electronic medical records, and multisite locations to support clinical trials and drug development. The Cleveland Clinic Lou Ruvo Center for Brain Health, for example, has four locations in the U.S. in an integrated trial network that can optimize AD patient recruitment and conduct of clinical trials.
The NIH is also responding to the crisis in drug development funding. The formation of the National Center for Advancing Translational Science (NCATS) is one milestone in reorganizing the NIH to orient more toward public–private partnerships and product development.Reference Collins63 NCATS has resources to support drug discovery and advance promising compounds through preclinical development, including assay development and high-throughput screening, synthesis, formulation, pharmacokinetics, toxicology, medicinal chemistry, molecular libraries probe production, genomics, interference RNA, tissue chips for drug screening, and technologies for identifying and validating drug targets. The Molecular Libraries Small Molecule Repository maintains a collection of >300,000 chemically diverse compounds for use in high throughput screening projects. The Rescuing and Repurposing Drugs program supports investigation of therapeutic effects of approved or abandoned compounds for indications other than the one originally intended. NCATS programs to support clinical stage development include the Clinical Translational Science Award (CTSA) funding a network of clinical trial sites throughout the nation and the Cures Acceleration Network (CAN). The National Institutes of Neurological Disease and Stroke (NINDS) operates a “virtual pharma” model of drug development, including bioactivity/efficacy studies, medicinal chemistry, pharmacokinetics, toxicology, manufacturing and formulation development, and phase 1 clinical trials for neurotherapeutic compounds as part of its Blueprint Neurotherapeutics Network.Reference Heemskerk, Farkas and Kaufmann64 The Neuroprotection Exploratory Trials in Parkinson's Disease (NET-PD)Reference Olanow, Wunderle and Kieburtz65, Reference Schneider, Elm and Parashos66 conducts clinical studies of neuroprotective compounds in Parkinson's disease, and the Network of Excellence in Neuroscience Clinical Trials (NeuroNext) is a network of trial sites organized by the NINDS to test drugs in adult and pediatric populations with neurological diseases. Observations made in Parkinson's disease trials may impact other neurodegenerative disorders, including AD. The National Institute on Aging (NIA) funds AD-related drug discovery (R21 grants) and development (UO1 grants), as well as the AD Cooperative Study (ADCS), is a multisite network for AD clinical trials, and the AD Neuroimaging Initiative (ADNI), which is a public–private partnership to study biomarkers in healthy elderly, those with mild cognitive impairment (MCI), and those with mild AD.Reference Cummings24, Reference Weiner, Veitch and Aisen67 The NIA also supports the National Alzheimer Coordinating Center (NACC), a database of standardized clinical and pathology data collected at NIA-funded Alzheimer Disease Centers.Reference Beekly, Ramos and van Belle68 The NIH Small Business Research (SBIR) funding program supports drug development in small businesses including biotechnology companies. Together, these programs provide a substantial federal resource for AD drug discovery and development. They comprise a broad platform for translational neuroscience in support of development of AD therapeutics.
Summary
The population of AD patients is rapidly expanding, and means of preventing or intervening in the disease process must be identified. Past approaches to drug development were effective in developing symptomatic agents, but they have consistently failed in the attempt to develop disease-modifying agents. New means of discovering agents and predicting human effects, better animal models, improved trial designs and outcomes, and more predictive biomarkers are needed. Translational neuroscience provides a framework for accelerating AD drug discovery and development.
Disclosures
Jeffery Cummings is a consultant for or has stocks/stock options in the following companies: ADAMAS, MedAvante, Neurokos, Neurotrax, Prana, QR, and Sonexa. He is a consultant for Abbott Laboratories, Acadia, Anavex, Astellas Pharma US, Avanir, Avid, Baxter, Bayer Healthcare, Bristol-Myers Squibb Co., Elan Pharmaceuticals, EnVivo, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Medtronic, Merck & Co, Inc., Neuronix, Otsuka Pharmaceuticals, Pain Therapeutics, Inc., Pfizer, Plexxicon, Sanofi Aventis, Signum Bioscience, Takeda, Toyama, and UBC. He performs consulting, teaching, and speaking work for Eisai, Forest Pharmaceuticals, Lundbeck, Novartis, and Ortho McNeil Janssen Scientific Affairs LLC. He owns the copyright for Neuropsychiatric Inventory. Ryan Walsh is a consultant/advisor for Lundbeck and a consultant, advisor, and member of the speaker's bureau for Teva. Kate Zhong is a consultant for Baycrest and Janssen Scientific Affairs. The remaining authors have nothing to disclose.







 Open access
Open access