Previously thought to be variants of multiple sclerosis, neuromyelitis optica spectrum disorders (NMOSDs) are now well recognized as distinct autoimmune CNS gliopathies. Reference Flanagan1 Diagnostic criteria from 2015 center around the occurrence of inflammatory lesions at one or more core clinical sites (including the optic nerves, spinal cord, and others), along with the presence of aquaporin-4 (AQP4)-IgG antibodies in serum, which target primarily astrocytes and initiate immune-mediated CNS damage. Reference Wingerchuk, Banwell and Bennett2 AQP4-seronegative patients need to meet more stringent clinical criteria to be classified as NMOSD. Approximately 1/4 to 1/3 of AQP4-seronegative patients are seropositive for myelin oligodendrocyte glycoprotein (MOG)-IgG antibodies, which find their antigen on oligodendrocytes. Reference Reindl and Waters3 Overall, clinical manifestations of MOG antibody disease (MOGAD) may resemble AQP4-positive NMOSD, but MOGAD has emerged as an entity with its own distinguishing features. Reference Weber, Derfuss, Metz and Brück4 Patients with NMOSD and MOGAD commonly experience neuropathic pain and headache as part of their illness. Reference Asseyer, Schmidt and Chien5 An observational study found that greater than 80% of 49 patients with NMOSD/MOGAD suffered from pain, including 5 of 12 MOGAD patients who experienced headache. Reference Asseyer, Schmidt and Chien5 These symptoms can also be the patient’s presenting concern, as we report here.
A 69-year-old right-handed man presented with 3 weeks of new-onset progressive, atraumatic headache. He described 10/10 “electric-shock”-like shooting pain radiating intermittently from the lateral neck up to the occiput and crown of the head. It was exacerbated by movement, but not by postural changes or Valsalva. He reported no migrainous features, no cranial autonomic symptoms, and no infectious symptoms. For the preceding 2 weeks, he also experienced gait ataxia, urinary hesitancy, and constipation. He reported no visual disturbance, bulbar dysfunction, sensory loss, or weakness. His medical history was notable for elevated body mass index, gastroesophageal reflux, and chronic erectile dysfunction. His only regular medication was rabeprazole. He was a former smoker of 40 pack-years and had no other pertinent exposures.
On examination, the patient was afebrile and hemodynamically stable. There was no nuchal rigidity, and his systemic examination was otherwise unremarkable. Cognition was intact. Afferent visual system and ocular fundus examinations were normal, as were the remaining cranial nerves. Motor exam showed bilateral arm and leg spasticity with preserved strength. Deep tendon reflexes were symmetric, with 2+ throughout, and 3+ at the brachioradialis and patellae bilaterally. He had no clonus and Hoffmann’s sign was absent bilaterally. Plantar responses were flexor. He reported allodynia in bilateral C2–C3 dermatomes. Vibratory sensation was diminished in both legs, returning to normal at the hips, without evidence of spinothalamic tract involvement. He exhibited mild bilateral dysmetria with finger-nose-finger testing and ambulated with a wide base. Romberg sign was positive.
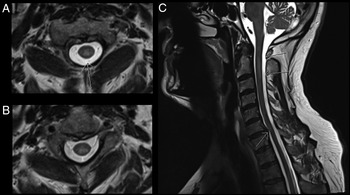
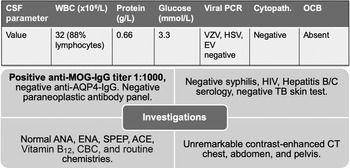
CT-CT angiogram of the head and neck revealed no vascular cause for the symptoms. MRI of the cervical spine with gadolinium showed a T2-hyperintense, intramedullary lesion in the high cervical spinal cord (Figure 1), enhancing on post-gadolinium images (not shown). Gadolinium-enhanced cranial MRI was normal. Unenhanced MRI of the thoracic spine and conus medullaris was also normal. Evaluation included serum, CSF, and body imaging studies (results in Figure 2). A serum cell-based immunofluorescence assay performed at Mitogen Advanced Diagnostic Laboratories (Calgary, Alberta, Canada) discovered a high-positive 1:1000 anti-MOG-IgG titer and was negative for anti-AQP4-IgG. This was confirmed by Mayo Clinic Laboratories (USA) serum flow cytometry cell-based testing with positive anti-MOG-IgG (titer 1:1000).
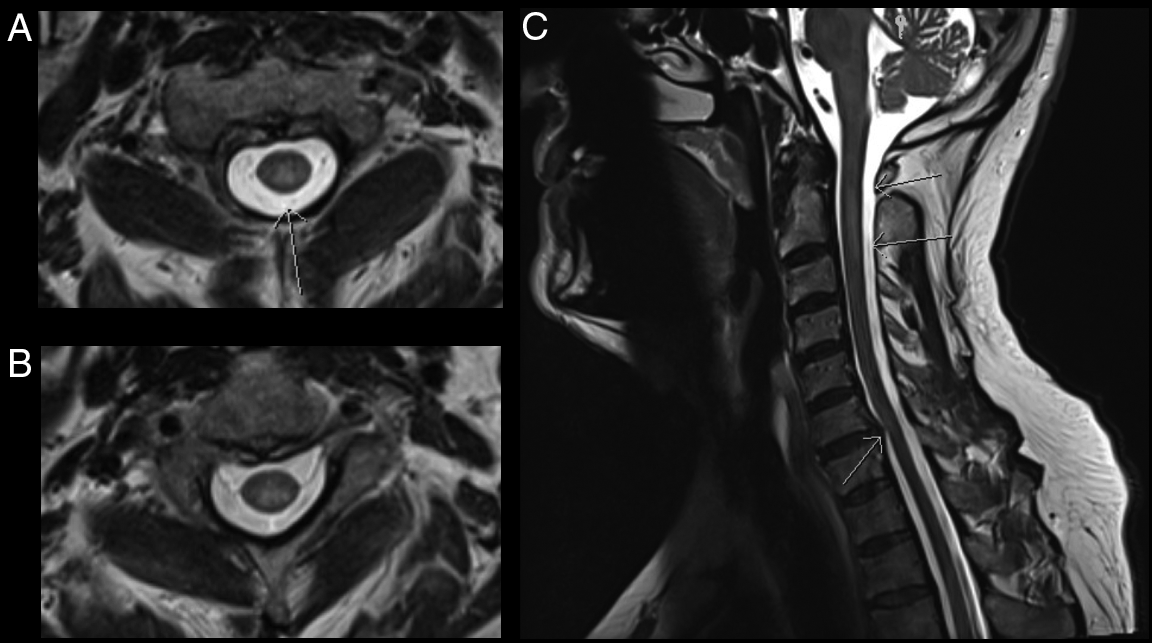
Figure 1: MRI C-spine axial images (A–B) demonstrate a T2-hyperintense, intramedullary cord lesion (arrow). On sagittal view (C) the lesion extends from C1–C2 interspace to the C3 vertebral body without evidence of spondylotic compression at these levels (upper arrows). Incidental posterior intervertebral disk bulge at C6–C7 without associated spinal cord signal change can also be appreciated (lower arrow).
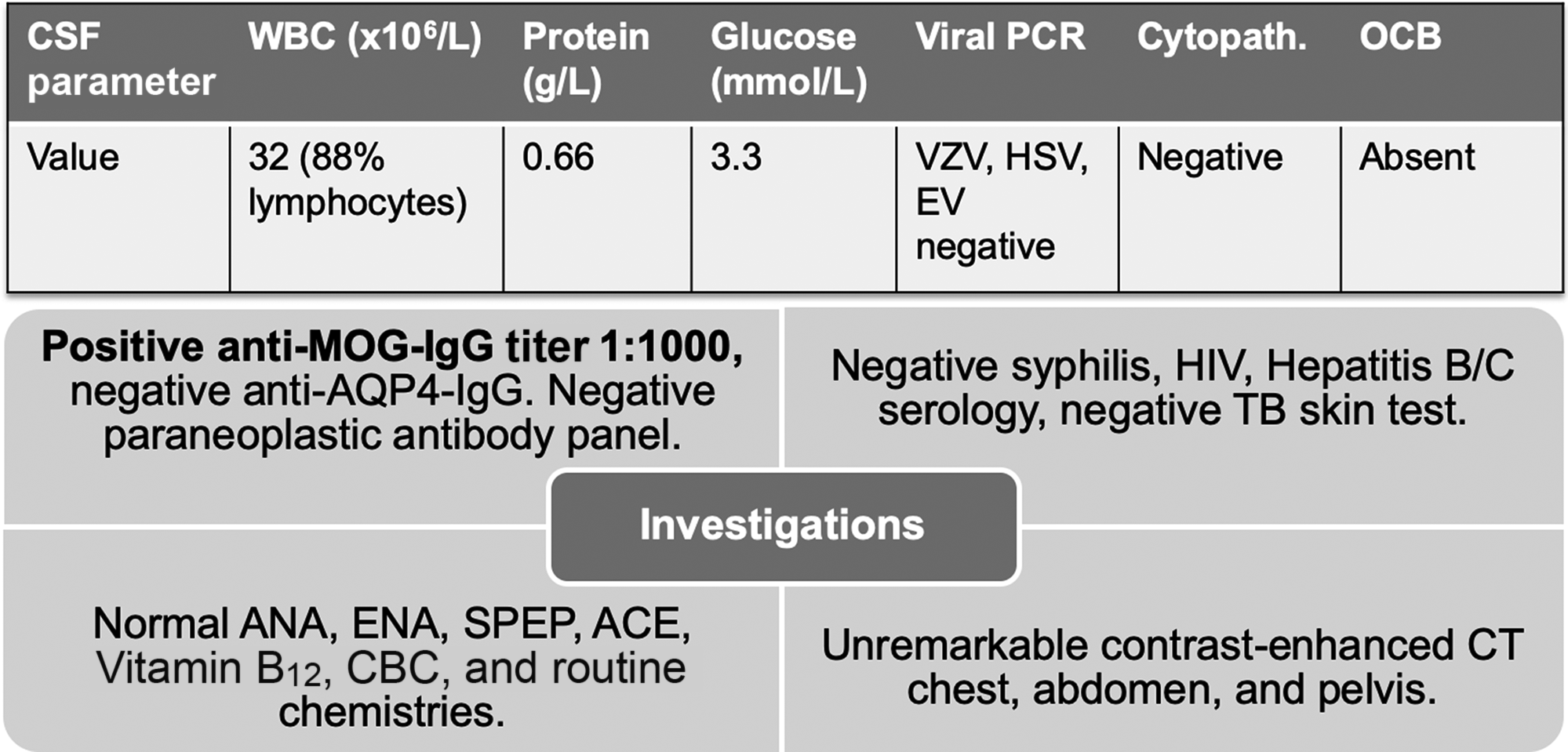
Figure 2: Summary of investigations. Anti-MOG-IgG and anti-AQP4-IgG testing was sent on serum only. Paraneoplastic panel tests were sent on both serum and CSF. Abbreviations: PCR = polymerase chain reaction; VZV = varicella zoster virus; HSV = herpes simplex virus; EV = enterovirus; Cytopath. = cytopathology; OCB = oligoclonal bands; TB = tuberculosis; ANA = anti-nuclear antibodies; ENA = extractable nuclear antigens; SPEP = serum protein electrophoresis; ACE = angiotensin-converting enzyme; MOG = myelin oligodendrocyte glycoprotein; AQP4 = aquaporin-4. Paraneoplastic antibody panel at Mitogen Advanced Diagnostic Laboratories tests for the following: Amphiphysin, Ri (NOVA-1), Yo, Hu, PNMA2 (Ma2/Ta), CV2.1, Recoverin, SOX1, Titin.
Treatment was initiated with 5 days of methylprednisolone 1 g intravenously daily, which resulted in rapid clinical and radiologic improvement. A 2-week taper of oral prednisone was prescribed at discharge, starting at a dose of 60 mg, decreasing by 10 mg every 3 days. A headache associated with ear and scalp tenderness, hand numbness, and chest tightness occurred 3 weeks following discharge, heralding a relapse which was treated by restarting oral prednisone with gradual tapering. A third relapse featuring symptoms of myelitis occurred off therapy approximately 8 months into the illness. Serum anti-MOG-IgG remained positive at 1 year (medium-positive; titer 1:100). The patient began long-term immunosuppression with azathioprine due to his recurrent symptoms.
A comprehensive evaluation determined that our patient’s symptoms were likely the result of MOGAD cervical myelitis. Consistent with this etiology, his symptoms and radiologic findings were responsive to corticosteroid treatment, and relapses occurred when they were weaned. The patient’s clinical onset occurred at an uncommonly advanced age. Of 50 MOG-IgG-seropositive patients with optic neuritis or myelitis, only 3 presented between 61–80 years of age with the median age of onset found to be 31 years (range 6–70). Reference Jarius, Ruprecht and Kleiter6
We hypothesize that our patient’s headache is attributable to spinal cord and possibly meningeal inflammation due to MOGAD. The pain was entirely responsive to treatment with steroids. Phenotypically, the headache resembles occipital neuralgia, as has been reported in other patients with high cervical myelitis. Reference Kissoon, Watson, Boes and Kantarci7 Allodynia was present in bilateral C2–C3 dermatomes, but unlike idiopathic occipital neuralgia, our patient experienced a more progressive, constant discomfort which was not reproduced by palpation over the occiput. Raised intracranial pressure (ICP) and aseptic meningitis have been documented in other patients with MOGAD; Reference Narayan, Wang, Sguigna, Husari and Greenberg8 however, clinicoradiologic features which usually accompany these entities were largely absent in our patient. Specifically, there was no worsening of the headache with recumbency or Valsalva, and optic disk edema was absent. A limitation is that we did not measure opening pressure during lumbar puncture to definitively exclude raised ICP. Recently, prodromal headaches reminiscent of migraine were reported in 49.6% of 129 MOG-IgG-seropositive optic neuritis patients, suspected to be related to peri-optic meningeal inflammation. Reference Asseyer, Hamblin and Messina9 Active myelitis was not a selection criterion for this cohort, and the headaches described differ in comparison to our patient, who had no classical migrainous features. Nevertheless, meningeal inflammation remains a putative headache contributor in our patient even though no leptomeningeal enhancement was detected using MRI. We speculate that the degree of meningeal inflammation in MOGAD exists on a continuum ranging from mild, imaging-occult disease to fulminant aseptic meningitis with raised ICP in some patients.
Pain negatively affects quality of life in patients with NMOSD and MOGAD and can be challenging to treat. Reference Asseyer, Schmidt and Chien5,Reference Qian, Lancia, Alvarez, Klawiter, Cross and Naismith10 Potential localizations for pain in these disorders span the CNS, from optic nerves to hypothalamus through the spinal cord. In addition to aforementioned headache mechanisms, pain may relate to the disruption of ascending nociceptive and descending antinociceptive pathways, disturbed excitatory–inhibitory balance, and central sensitization. Reference Bradl, Kanamori and Nakashima11 Further prospective study of symptom management in NMOSD and MOGAD is needed, since not all patients will have resolution of their symptoms with immunosuppression.
Acknowledgments
We thank the patient for providing informed consent to publish this case report.
Conflict of Interest
No conflicts of interest are declared by the authors.
Statement of Authorship
JDK, SEJ, and KA were involved in clinical care of the patient. JDK wrote the first draft of the manuscript and prepared the figures. SEJ and KA participated in manuscript revision and production of the final draft.