Introduction
The genus Vanilla Plumier ex Miller is included into the Orchidaceae family within monocots and contains about 130 species mostly wild, among which 30 have been reported to wear aromatic fruits (Soto-Arenas, Reference Soto-Arenas1999). However, only the widely cultivated and commercial Vanilla planifolia Andrews and the far less important V. X tahitensis J. W. Moore presents commercial interest (Lepers-Andrzejewski et al., Reference Lepers-Andrzejewski, Causse, Caromel, Wong and Dron2012). Other species such as V. pompona Schiede and V. odorata C. Presl. exist within vanilla plantations or are harvested in the wild but are not economically important (Soto-Arenas and Cameron, Reference Soto-Arenas and Cameron2003).
Cultivated vanilla around the world arose from the domestication of a unique subpopulation of V. planifolia from Meso America (Bory et al., Reference Bory, Lubinsky, Risterucci, Noyer, Grisoni, Duval and Besse2008a; Lubinsky et al., Reference Lubinsky, Bory, Hernández Hernández, Kim and Gómez-Pompa2008; Minoo et al., Reference Minoo, Jayakumar, Veena, Vimala, Basha, Saji, Nirmal and Peter2008; Verma et al., Reference Verma, Chakrabarty, Jena, Mishra, Singh, Sawant and Tuli2009). As a result of this domestication process, commercial vanilla exhibits very limited genetic diversity (Bory et al., Reference Bory, Da Silva, Risterucci, Grisoni, Besse and Duval2008b; Minoo et al., Reference Minoo, Jayakumar, Veena, Vimala, Basha, Saji, Nirmal and Peter2008). Furthermore, the prevalent practice of asexual propagation contributes to the genetic uniformity of this crop, rendering it more susceptible to disease infection. Besides, obtaining vanilla with high vanillin content and disease resistance is challenging. Breeding programmes are limited by (i) the few available germplasm resource databases; (ii) the asexual propagation commonly used by farmers; (iii) the complexity of the genome (Brown et al., Reference Brown, Bourge, Maunoury, Wong, Bianchi, Lepers-Andrzejewski, Besse, Siljak-Yakovlev, Dron and Satiat-Jeunemaître2017); and (iv) the relatively long-life cycle of vanilla. In fact, the first flowers only appear around the third year of cuttings and by the seventh year after seed germination.
Regardless of the high economic value of vanilla (the second most expensive spice after saffron), few breeding efforts have been made (Lepers-Andrzejewski et al., Reference Lepers-Andrzejewski, Causse, Caromel, Wong and Dron2012; Chambers, Reference Chambers2018). One of them was conducted in Madagascar, dating back to the middle of the twentieth century (1944; Tonnier, Reference Tonnier1960; Dequaire, Reference Dequaire1976). In view of producing disease-resistant and high vanillin content varieties, several crossing strategies were conducted between introduced, local, cultivated and wild species. The improvement aimed to keep the potential traits of V. planifolia, especially the aroma profile, and thus used this species as a basis in different crossing configurations and mainly attempted to introduce the resistance and hardiness traits from other species such as V. phaeantha Rchb. F., V. pompona Schiede, V. madagascariensis Rolfe, V. abundiflora J. J. Sm., V. coursii H. Perrier, V. françoisii H. Perrier, V. walkeriae Wight and V. zanzibarica Rolf . Additionally, V. X tahitensis J.W. Moore was also used to introduce the bean indehiscence trait into V. planifolia. The intra- and inter-specific mating and trait selection led to a thousand hybrids from which Tsitaitra and ‘Magnitry Ampotony’, reported as hybrids of (V. planifolia x V. pompona) V. planifolia and V. planifolia x V. X tahitensis cv Haapape, were retained for disease resistance and vanillin content, respectively (Grisoni and Nany, Reference Grisoni and Nany2021). Unfortunately, the programme was abandoned in 2000, about 50 years after its beginning and the genetic resources seem to have been dispersed or lost and the programme is poorly documented. Since then and as far as we know, no research has been conducted on the ancient breeding programme. It is, therefore, urgent to assess the remaining resources for the conservation of the breeding materials and for future genetic improvement. In light of the growing threats to genetic resources loss in crop species (Rao, Reference Rao2004), including vanilla (Householder et al., Reference Householder, Janovec, Mozambite, Maceda, Wells, Valega and Christenson2010; Herrera-Cabrera et al., Reference Herrera-Cabrera, Salazar-Rojas, Delgado-Alvarado, Contreras, Contreras and Cervantes-Vargas2012; Hu et al., Reference Hu, Resende, Bombarely, Brym, Bassil and Chambers2019) and the increasing challenges faced by agricultural systems, comprehensive assessments of these genetic resources are crucial (Govindaraj et al., Reference Govindaraj, Vetriventhan and Srinivasan2015). Understanding the genetic makeup of the resources allows us to prioritize conservation efforts, develop conservation plans and utilize the genetic potential effectively. By assessing the genetic diversity and population structure of these resources, we can identify and conserve unique and rare genetic traits, which may hold significant value for developing improved vanilla varieties. To fill this gap, the present study aims to investigate the genetic diversity and structure of the genotypes kept within a local collection in Antalaha, in the SAVA region, in the north-eastern part of Madagascar. The vanilla collection was established in Antalaha by Ramanandraibe Exportation Société Anonyme, known as Rama Export (a local firm exporting vanilla), and containing a number of genotypes mainly collected from the abandoned vanilla research station with rare plants from other places within the SAVA region.
Among molecular techniques, next-generation sequencing (NGS) has become a popular practice in conservation programmes for diversity investigation (Ekblom and Galindo, Reference Ekblom and Galindo2011; Hunter et al., Reference Hunter, Hoban, Bruford, Segelbacher and Bernatchez2018). NGS is a high-throughput genotyping that allows sequencing of several individuals at once for a reasonable time (Kumar et al., Reference Kumar, Banks and Cloutier2012; Shirasawa et al., Reference Shirasawa, Hirakawa and Isobe2016). Due to their cost-effectiveness and flexibility, genotyping-by-sequencing (GBS) and restriction associated-site (RAD) protocols are now very attractive NGS methods. Double digest RAD-seq (ddRAD-seq) is one of the several short-read library sequencing (SRSL) within the RAD family that targets homologous regions across the genome and relies on two restriction enzymes. NGS protocol was used in the present study since it has been successfully used in several crop species having complex genomes like vanilla (Saintenac et al., Reference Saintenac, Jiang, Wang and Akhunov2013; Feng et al., Reference Feng, Zhao, Li, Zhang, Qu, Li, Li, Lin and Pu2020; Natarajan et al., Reference Natarajan, Hossain, Kim, Denison, Ferdous, Jung, Park and Nou2020) as well as in previous studies specifically focused on vanilla genomics (Hu et al., Reference Hu, Resende, Bombarely, Brym, Bassil and Chambers2019; Chambers et al., Reference Chambers, Cibrián-Jaramillo, Karremans, Martinez, Hernandez-Hernandez and Brym2021; Favre et al., Reference Favre, Jourda, Grisoni, Piet, Rivallan, Dijoux and Charron2022).
Materials and methods
Plant material
Fifty-six vanilla plants maintained in the Rama Export collection in Antalaha, SAVA region, Madagascar were used in this study. The germplasm panel includes remaining progenitors and hybrids from the ancient breeding programme (in Ambohitsara, Antalaha) pertaining to five groups easily identified phenotypically: V. planifolia, V. pompona, hybrids labelled as Tsitaitra and other varieties referred to as Tsivaky and Vanille Banane (Fig. 1). The variety Tsitaitra has been documented as a result of (V. planifolia x V. pompona) V. planifolia crossing (Grisoni and Nany, Reference Grisoni and Nany2021). However, information regarding Tsivaky and Vanille Banane is missing.

Figure 1. Morphological differences between the five phenotypic groups. (a) Vanilla planifolia; (b) Tsitaitra; (c) Vanilla pompona; (d) Tsivaky; (e) Vanille Banane.
ddRAD-seq data generation
To assess genetic diversity and structure of the studied accessions, we employed the ddRAD-seq approach (Peterson et al., Reference Peterson, Weber, Kay, Fisher and Hoekstra2012) to generate sequences data. Total genomic DNA was isolated using Doyle and Doyle's CTAB-based (cetyltrimethylammonium bromide) protocol (Reference Doyle and Doyle1987) with some modifications. Briefly, to release the cell content, 400 mg of leaf were crushed in 1 ml of CTAB buffer solution preheated to 65 ° C (0.1 M Tris pH8, 20 mM EDTA pH8.5, 1.4 M NaCl, 2% CTAB, 0.5 mM Na2S2O5). β-mercaptoethanol and PVP 1% each were added right before extraction. The solution was then incubated at 65 ° C for 45 min with shaking every 5 min. DNA was separated from cell debris using chloroform/isoamyl alcohol (24:1, v/v) followed by centrifugation. This step was repeated twice to avoid contamination. The DNA in solution was subsequently precipitated with 3 M sodium acetate, pH 5.2 (1/10 volume) and 2/3 volume isopropanol stored at room temperature. Then, the DNA pellet was washed twice with cold 70% ethanol. After evaporation of residual alcohol, DNA was resuspended in 30 μl of TE1x solution mixed with RNase A DNAse free to remove molecular RNAs. Finally, the DNAs were stored at −20 °C.
Libraries were prepared following the protocol described by Peterson et al. (Reference Peterson, Weber, Kay, Fisher and Hoekstra2012). Prior library preparation, DNA integrity was first evaluated on 1% agarose gel electrophoresis and quantity was assessed with Qubit (2.0 Fluorometer, Invitrogen, Carlsbad, CA, USA). These tasks were performed to ensure the success of the DNA sequencing as DNA degradation can affect the accuracy of the data. In addition, libraries require a minimum amount of DNA to ensure sufficient coverage and accuracy during sequencing. In order to avoid sequencing bias trough over- or underrepresentation of samples, DNA concentration was then normalized to 600 ng in 30 μl for each sample. For each individual sample, genomic DNA was digested with two restriction enzymes ECORI HF (NEB) and NLAIII (NEB) by incubating at 37 °C for 18 h. Digestion success was assessed on 1% of agarose gel. Then 200 ng of double digested DNA were ligated with two adaptors at each restriction cut site using T4 DNA ligase (NEB). Both adaptors are designed to be compatible with each cut site overhang and serve for later amplification step as they are complementary to the Illumina PCR oligo-sequences. The first adaptor is barcoded, the identifier is specific for each individual to allow to identify reads from each sample during data processing. The second adaptor is common for all individual in the same library. The barcoded samples in each set were then pooled together. To sequence short representative DNA fragments, we selected adaptor-ligated fragments ranging from 300 to 600 bp. DNA fragment size selection was performed at the Genomics Core in Leuven, Belgium. A specific index was added by PCR at the common adaptor side of size selected fragments. The index allows to pool together two or more sets of individuals. The prepared libraries were sent to GENEWIZ (Steiblingen, Germany) company for sequencing using Illumina NovaSeq 250 bp length paired end sequencing platform.
Read quality assessment, cleaning and demultiplexing; alignment to the reference genome
Raw sequence reads were demultiplexed with Stacks v2.53 (Catchen et al., Reference Catchen, Amores, Hohenlohe, Cresko and Postlethwait J2011) using an individual's dual index-barcode information. Read quality was then checked using Fastqc v0.11.9 (Andrews et al., Reference Andrews, Krueger, Seconds-Pichon, Biggins and Wingett2014). To retain only fragments of good quality, reads were trimmed from 220 bases and those with uncalled base or base quality lower than 30 were discarded.
Paired end reads were aligned to the published reference genome (Hasing et al., Reference Hasing, Tang, Brym, Khazi, Huang and Chambers2020) widely used in similar studies of vanilla (Chambers et al., Reference Chambers, Cibrián-Jaramillo, Karremans, Martinez, Hernandez-Hernandez and Brym2021; Ellestad et al., Reference Ellestad, Pérez-Farrera and Buerki2022; Favre et al., Reference Favre, Jourda, Grisoni, Piet, Rivallan, Dijoux and Charron2022) using the Burrows-Wheeler Aligner (BWA) v0.7.15 (Li and Durbin, Reference Li and Durbin2009). The BWA mem function was run with its default parameters as described by (Rochette and Catchen, Reference Rochette and Catchen2017). SAMtools v.1.6 (Danecek et al., Reference Danecek, Bonfield, Liddle, Marshall, Ohan, Pollard, Whitwham, Keane, McCarthy, Davies and Li2021) was used to convert the aligned reads from SAM to BAM format and sort the reads by position for variant calling.
SNP calling and filtering
Various tools have been designed to call variants from wide-genome sequencing. They primarily vary in the algorithms employed for genotype estimation, which can result in varying outputs with distinct levels of overall accuracy (Pirooznia et al., Reference Pirooznia, Kramer and Parla2014; Torkamaneh et al., Reference Torkamaneh, Laroche and Belzile2016). Some of them may be prone to bias leading to potentially unreliable results (Stift et al., Reference Stift, Kolář and Meirmans2019). To ensure that only high-quality SNPs are retained for the germplasm characterization, common SNPs detected by the three pipelines widely used for SRSL genotyping, i.e. Stacks (Catchen et al., Reference Catchen, Hohenlohe, Bassham, Amores and Cresko2013), BCFtools (Danecek et al., Reference Danecek, Bonfield, Liddle, Marshall, Ohan, Pollard, Whitwham, Keane, McCarthy, Davies and Li2021, https://samtools.github.io/bcftools/bcftools.html) and GATK (Poplin et al., Reference Poplin, Ruano-Rubio, De Pristo, Fennell, Carneiro, Van der Auwera and Banks2017), were selected and used for downstream analysis.
Only reads with a mapping quality greater than Q30 were used for genotyping. The SNPs were further filtered according to the following criteria: present in at least 80% of individuals; minimum allele frequency of 0.01, minimum and maximum depth for a site: 10 and 250, respectively; minimum and maximum depth for a genotype per individual: 10 and 250, respectively. This task was performed by VCFtools v4.2 (Danecek et al., Reference Danecek, Auton, Abecasis, Albers, Banks, DePristo, Handsaker, Lunter, Marth, Sherry, McVean and Durbin2011), which was again used to identify common SNPs between pipelines. Consecutive SNPs within 10 bp window were excluded, resulting in a refined set of SNPs used for genetic diversity and structure analysis. Our preliminary analysis revealed that the vanilla population under study exhibited a relatively low level of linkage disequilibrium (LD), consistent with the findings of Ellestad et al. (Reference Ellestad, Pérez-Farrera and Buerki2022), which demonstrated that LD had no significant impact on clustering results. Therefore, instead of considering LD, our focus was on filtering successive SNPs to efficiently capture the overall genetic variation across the genome. SNP location and function were identified with VEP (McLaren et al., Reference McLaren, Gil, Hunt, Riat, Ritchie, Thormann, Flicek and Cunningham2016) to further validate the SNPs.
Additionally, a comprehensive evaluation of each tool's performance was conducted to determine the most suitable approach, with a specific emphasis on the accuracy and reliability of vanilla SNP genotyping. This evaluation was carried out to provide recommendations that would facilitate the study of vanilla genetic diversity. This evaluation encompassed several factors, including CPU time, number of called SNPs, missing data, loci coverage and heterozygosity. CPU time represents the actual processing time executed by the CPU to complete assigned tasks.
Basic statistics of genetic variation and population structure
Genetic diversity and clustering were analysed using the concordant and common filtered SNPs detected by the three pipelines. To assess the level of genetic variation within the population, the expected heterozygosity (He) and observed heterozygosity (Ho) were computed. These parameters provide insights into the genetic diversity among accessions. Additionally, the inbreeding coefficient (Fis) and identified private alleles (Pa) were calculated to further characterize the population. The computations for these parameters were performed using GAD v.1.1 (Lewis and Zaykin, Reference Lewis and Zaykin2002). To identify shared alleles between the known hybrid Tsitaitra and its documented parents (V. planifolia and V. pompona), BCFtools was employed (Danecek et al., Reference Danecek, Bonfield, Liddle, Marshall, Ohan, Pollard, Whitwham, Keane, McCarthy, Davies and Li2021). Additionally, an analysis was conducted to determine the potential parents of the other presumed hybrids named Tsivaky.
ADMIXTURE v1.3 (Alexander et al., Reference Alexander, Shringarpure, Novembre and Lange2015), a model-based cluster estimation, with K ranging from 1 to 10 and 1000 bootstrap replicates, was used to identify genetically homogeneous groups within the population. Cross-validation (CV) values were used to identify the appropriate K-value. The tool evaluates the fit of different clustering scenarios and the results were summarized by simple graphs of K, where the lowest peak indicates the theoretically best number of groups. Principal component analysis (PCA) and unweighted pair group method with arithmetic mean (UPGMA) tree (based on Identity by State distance) were also used to visualize genetic relationships between accessions using TASSEL v5 (Bradbury et al., Reference Bradbury, Zhang, Kroon, Casstevens, Ramdoss and Buckler2007) and ggplot2 of R v4.2.0 (Kronthaler and Zöllner, Reference Kronthaler and Zöllner2021) for plotting. The UPGMA approach served as complementary information to PCA and ADMIXTURE analyses. It provided a hierarchical representation of the population structure, allowing the identification of genetic subgroups and organize individuals into clusters based on their genetic similarity.
Results
ddRAD sequencing
A total of 865 million reads were generated from Illumina NovaSeq sequencing of the 56 vanilla samples. After cleaning and filtering of the raw sequence data, 72.5% of the reads were retained. On average, each sample yielded approximately 11 million cleaned and high-quality reads (Q > 30). To ensure data integrity, eleven individuals with a high rate of missing data were excluded from downstream analysis, resulting in a final cohort of 45 individuals (Table S1). The genome-wide GC content of the studied samples determined to be was 34%, a value falling within the expected range for monocots, as estimated by Šmarda et al. (Reference Šmarda, Bureš, Horová, Leitch, Mucina, Pacini, Tichý, Grulich and Rotreklová2014) to be between 33.6 and 48.9%. Furthermore, the observed GC content is slightly higher compared to the previously reported value of 30.8% for V. planifolia by Hasing et al. (Reference Hasing, Tang, Brym, Khazi, Huang and Chambers2020). An average of 99.3% (SD 1.0) of total reads was physically mapped to the reference genome.
Common SNP discovery
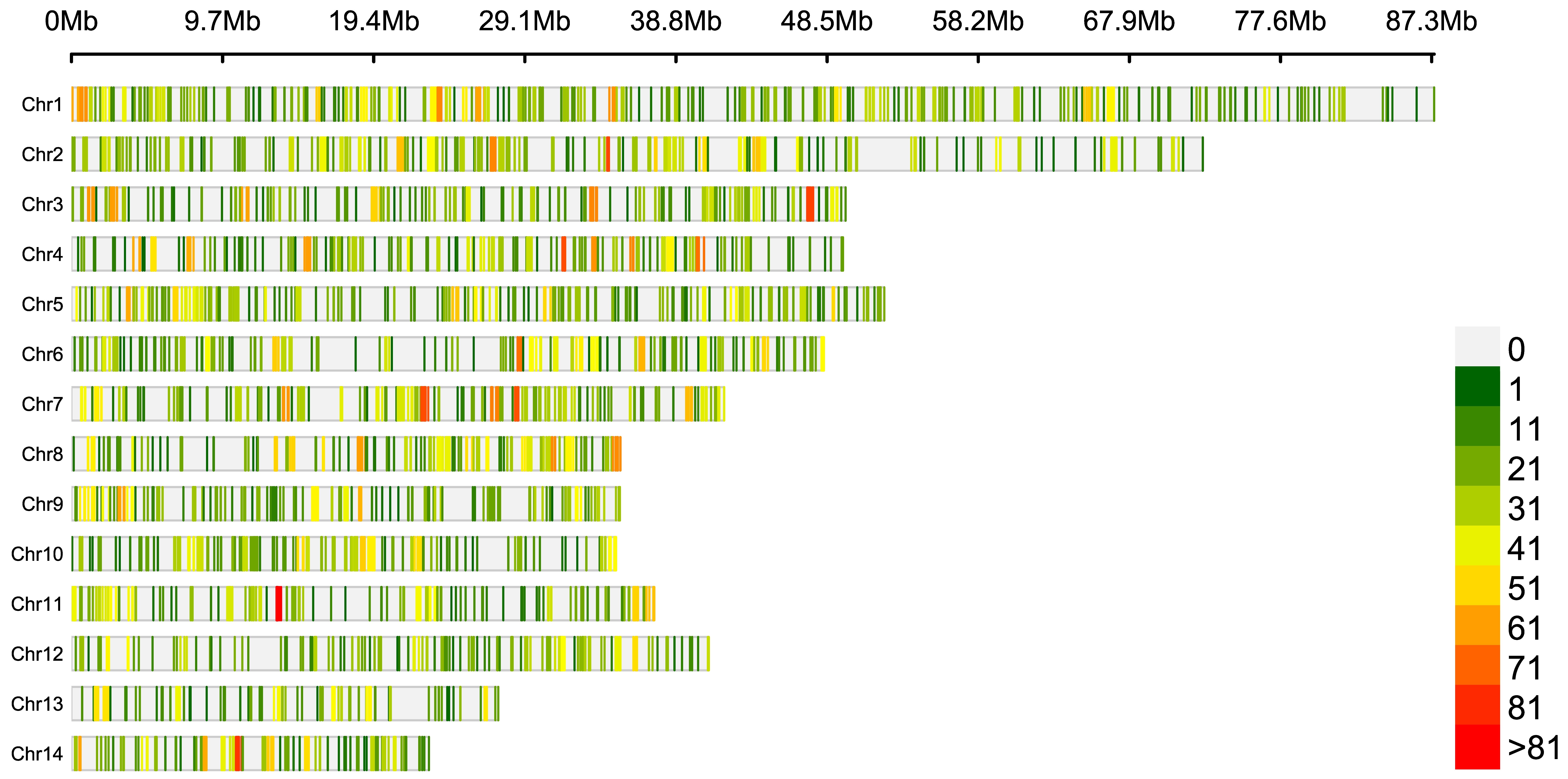
The 23,701 concordant common and high-quality SNPs were physically mapped to the genome, which were proportionally distributed across the 14 vanilla chromosomes (Hasing et al., Reference Hasing, Tang, Brym, Khazi, Huang and Chambers2020), i.e. the per-chromosome number of SNPs appeared to be proportional to its size (Figure S1). A total of 2998 of these variants were located in coding regions (Table S2).
Genome-wide genetic variation
Using the 23,701 common SNPs, the revealed genetic diversity of the studied vanilla accessions was high, with an observed heterozygosity (Ho) of 0.39. Notably, V. pompona and the hybrid Tsitaitra group presented the highest values (0.50 and 0.47), while V. planifolia exhibited the lowest value (0.26). The occurrence of private alleles (alleles that occur only within a given group) was noticed in each phenotypic group, with the largest being observed within the Tsivaky phenotype (2866). Negative Fis was obtained for all groups suggesting an excess of heterozygosity (Table 1), as previously reported for vegetatively propagated plants are (Elias et al., Reference Elias, Penet, Vindry, McKey, Panaud and Robert2001; Ge et al., Reference Ge, Liu, Wang, Schaal and Chiang2005), including vanilla (Hu et al., Reference Hu, Resende, Bombarely, Brym, Bassil and Chambers2019).
Table 1. Descriptive statistics on ddRAD sequencing and the genetic diversity of the 45 studied vanilla accessions from the 23,701 common SNPs

N, number of individuals; He, expected heterozygosity; Ho, observed heterozygosity; Fis, inbreeding coefficient; Pa, private alleles.
Population structure and genetic relationships
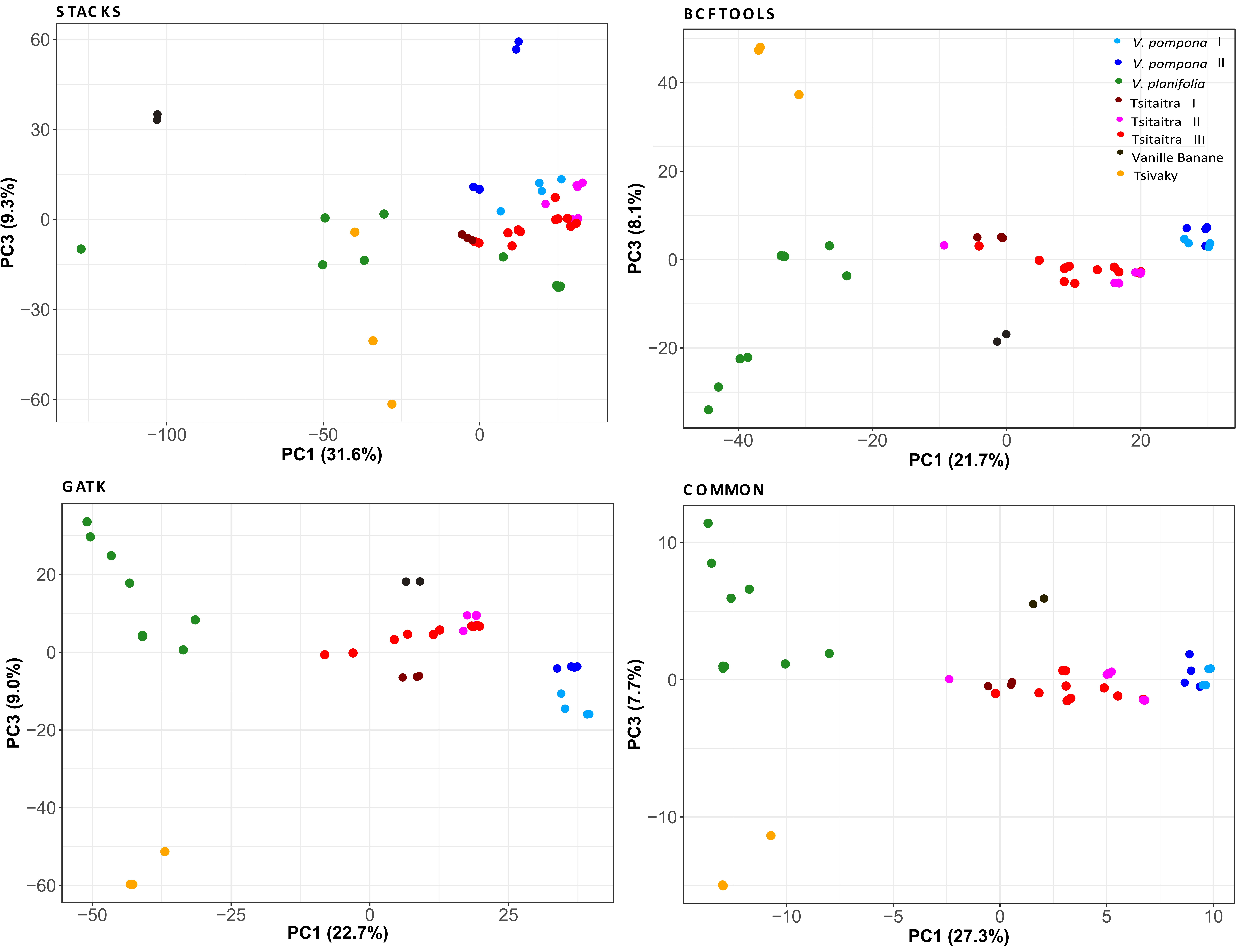
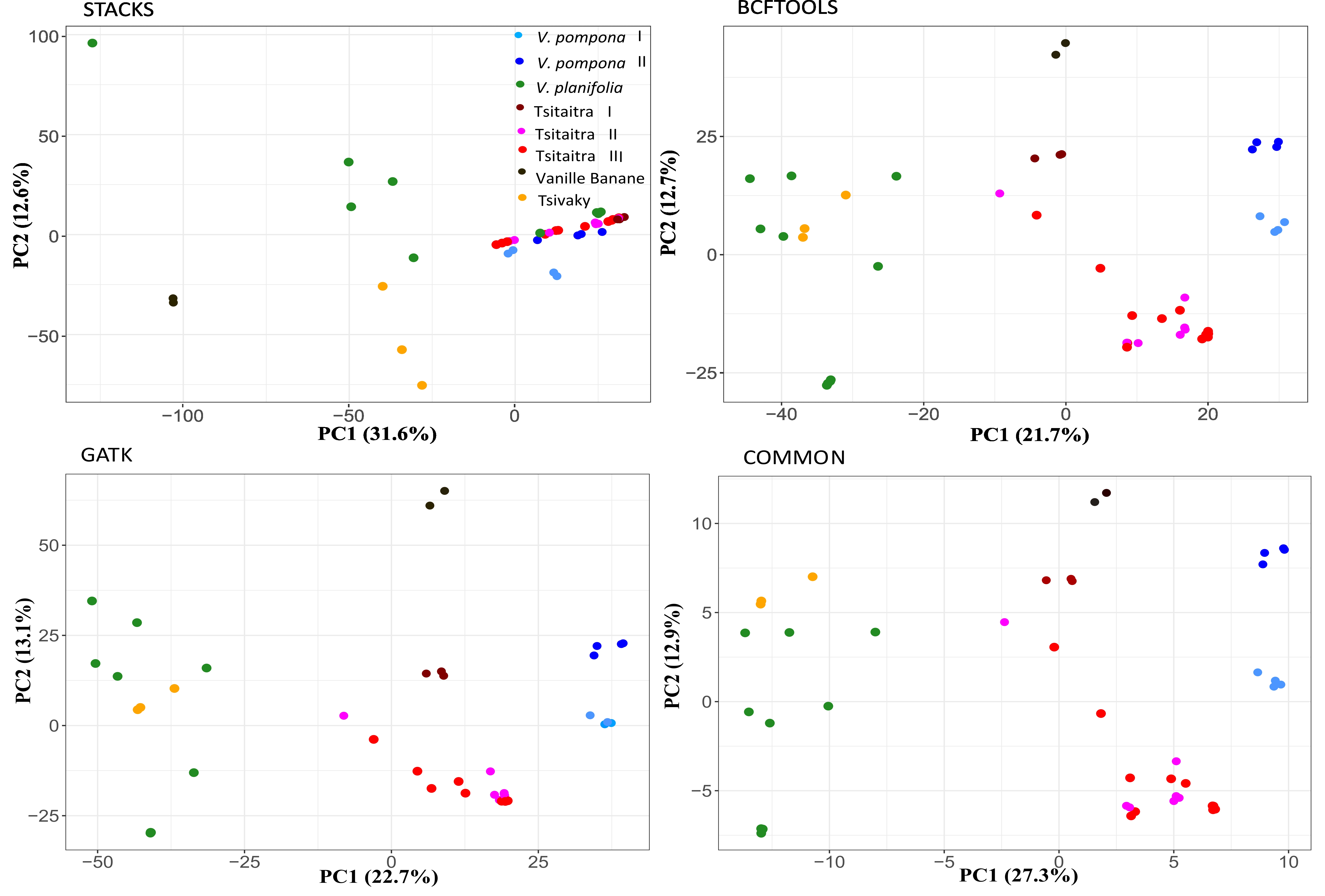
The genetic structure of the 45 vanilla accessions was analysed using PCA, UPGMA and ADMIXTURE based on the 23,701 detected common SNPs by the tree used pipelines. The results revealed distinct separation among the five phenotypic groups. Regarding the PCA results, PC 1 successfully separated three major groups: one consisting of V. pompona accessions, another containing both Tsitaitra and Vanille Banane and the last one comprising both V. planifolia and Tsivaky. PC 2 further distinguished Tsitaitra from and Vanille Banane (Fig. 2) and PC 3 separated V. planifolia from Tsivaky (Figure S2).

Figure 2. First and second principal components of the PCA of the 45 vanilla samples with the filtered SNPs from the 23,701common SNPs detected by all three protocols together.
UPGMA tree also demonstrated clear separation of the five phenotypic groups, with Vanille Banane appearing as the most distinct group (Fig. 3). Interestingly, within V. pompona and Tsitaitra, the analysis revealed the presence of subgroups. Specifically, two subgroups were observed within V. pompona (V. pompona I and II), while three subgroups were identified within Tsitaitra (Tsitaitra I, II and III). PCA further confirmed the distinction between the two subgroups of V. pompona, although the subgroups of Tsitaitra appeared less distinct on the PCA plot, with Tsitaitra II and III showing some overlap (Fig. 2).

Figure 3. UPGMA tree of the 45 individuals based on Identity by State distance from the 23,701 common loci detected by Stacks, BCFtools and GATK together.
The analysis of ADMIXTURE results revealed that the optimal K-value detected by the CV error plot was K = 7 (Fig. 4). This finding was consistent with the outcomes obtained from PCA (Fig. 2) and UPGMA (Fig. 3) analyses. Indeed, when K = 7 was applied, the five phenotypic groups were clearly segregated from each other (Fig. 5), confirming the concordance among the different analytical approaches. Within V. pompona and Tsitaitra the presence of subgroups was evident, with one exception: specifically, all subgroups within V. pomponas and Tsitaitra were successfully differentiated, except for Tsitaitra I, which appeared indistinguishable from V. pompona II in the analysis (Fig. 5, k = 7), consistently to the finding from UPGMA (Fig. 3) where both Tsitaitra I and V. pompona seem to be genetically close to each other.

Figure 4. Cross validation (CV) error plot, the lowest value indicates the appropriate number of populations (K = 7).

Figure 5. Genetic structure of the 45 cultivated vanilla accessions revealed by ADMIXTURE with the 23,701 common SNPs detected by Stacks, BCFtools and GATK. Pom (V. pompona), Pla (V. planifolia), Tsit (Tsitaitra), V.Ban (Vanille Banane), Tsiv (Tsivaky).
Parentage of putative hybrids
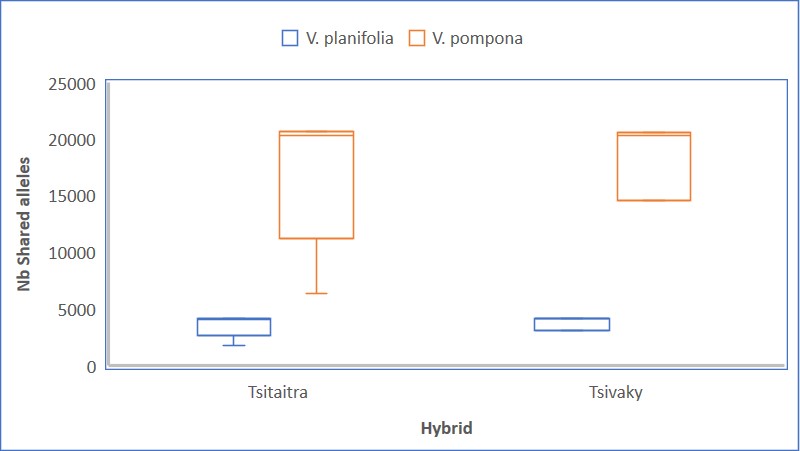
In the investigation of hybrid types and their parentage, the parentage of the hybrid Tsitaitra was confirmed to be a hybrid of V. planifolia and V. pompona through the identification of shared alleles (Figure S4). Among the three subgroups identified within Tsitaitra based on the targeted genome regions in the study, subtle differences in shared alleles with the presumed parents were observed. Group I exhibited the highest number of shared alleles with V. pompona (92.2%), while groups II and III displayed similar numbers of shared alleles (87.2% for group II and 87.3% for group III).
Regarding the analysis of Tsivaky, for which the parents are completely unknown, the same analysis was conducted using V. planifolia and V. pompona as putative parents. The results showed discrepancies between the findings from the PCA plot and the shared alleles analysis (Figure S3). While the PCA plot indicated a closer relationship between Tsivaky and V. planifolia, the shared alleles analysis suggested a closer relationship between Tsivaky and V. pompona rather than with V. planifolia. These differences can be explained by several hypotheses, which are further discussed below.
Comparison of the used genotyping tools (Stacks, BCFtools and GATK)
In terms of SNP genotyping, Stacks exhibited the highest SNP count, detecting a total of 316,398 variants. BCFtools and GATK, on the other hand, provided a number of markers quite closer to each other, with 80,888 and 126,391 respectively. The missing data rates were comparable among the pipelines; although Stacks had a greater value than the other two. Interestingly, Stacks showed the lowest loci coverage value (33×), while BCFtools and GATK displayed higher sequencing coverage (64.6×, 51.4×). Additionally, Stacks appeared to call for less heterozygous genotypes compared to the other pipelines (Table S3).
Regarding population structure analysis, BCFtools and GATK provided comparable outputs, which were consistent with the SNPs commonly detected by all three pipelines mentioned above. However, when using Stacks, the phenotypic groups showed a mixed distribution, except for the Vanille Banane group (Figure S3 for the first and second components, and Figure S2 for the first and third components).
Discussion
Understanding the genetic diversity and the structure of ex-situ populations is crucial for better management of resources and for genetic improvement programmes (Allendorf et al., Reference Allendorf, Hohenlohe and Luikart2010). Furthermore, for conservation purposes, comprehensive knowledge of germplasm for understanding the survival of germplasm and growth pattern becomes possible (Pillon et al., Reference Pillon, Qamaruz-Zaman, Fay, Hendoux and Piquot2007). The analysis of the 23,701 genome-wide genotyped SNPs provided valuable insights into the genetic differentiation among the five distinct phenotypic groups. Moreover, the presence of subgroups within certain phenotypes/species was also observed, adding another layer of complexity to the population structure of vanilla within the collection.
The five phenotypic groups were segregated by SNP data
The analysis of genetic diversity using ddRAD sequencing revealed robust clustering and distinct separation among the five phenotypic groups: V. planifolia, V. pompona, Tsitaitra Tsivaky and Vanille Banane. This clear separation was evident through the three complementary analytical approaches, including PCA (Fig. 2), UPGMA (Fig. 3) and ADMIXTURE (Fig. 5). The UPGMA tree analysis further emphasized the distinctness of the five phenotypic groups, with Vanille Banane appearing as the most distinct group (Fig. 3). This distinctiveness raises the possibility that Vanille Banane may represent another vanilla species. Moreover, the sterility observed in Vanille Banane suggests a potential polyploid or aneuploid nature, which could result from ancestors that are not represented in the current study. The inclusion of additional accessions from different regions, including known accessions, in future research will provide valuable insights into the origins and characteristics of Vanille Banane and contribute to a comprehensive understanding of vanilla diversity.
Regarding the genetic diversity, among the accessions, the highest observed heterozygosity was found for V. pompona (0.50) while V. planifolia had a much lower value (0.26). This is fully consistent with the previously reported low genetic diversity of V. planifolia (Duval et al., Reference Duval, Bory, Andrzejewski, Grisoni, Messe, Causse, Charon, Dron, Odoux and Wong2006; Sreedhar et al., Reference Sreedhar, Venkatachalam, Roohie and Bhagyalakshmi2007; Bory et al., Reference Bory, Catrice, Brown, Leitch, Gigant, Chiroleu, Grisoni, Duval and Besse2008c; Minoo et al., Reference Minoo, Jayakumar, Veena, Vimala, Basha, Saji, Nirmal and Peter2008). Intense selection on a specific trait, particularly the aroma profile, may result in the loss of polymorphism on certain loci. High heterozygosity was obtained from the hybrid individuals (Tsitaitra, 0.47) compared to that of V. planifolia (026). Furthermore, a substantial number of specific alleles were identified within each group (Table 1), indicating the presence of a diverse gene pool that holds potential for selecting and developing desired traits in future vanilla breeding.
Revealing subgroups within V. pompona and the hybrid Tsitaitra
Within the V. pompona accessions and Tsitaitra, our analysis revealed the presence of respectively, two and three distinct subgroups (Fig. 3). Regarding the V. pompona, genetic separation among accessions from different geographical origins has been previously reported (Bory et al., Reference Bory, Lubinsky, Risterucci, Noyer, Grisoni, Duval and Besse2008a). This finding could result from the origin of introduced V. pompona progenitors in Madagascar, that raised from different geographical areas (West Indies, France, Comoros, Mauritius and Moye Bampamperu (Grisoni and Nany, Reference Grisoni and Nany2021). The segregation of accessions within the Tsitaitra group may be attributed to various crossbreeding configurations implemented during the breeding programme (personal communication with a former technician involved in the programme). Tsitaitra is a very robust vanilla vine, resistant to Fusarium sp. and to challenging environmental condition. In general, this variety shows thick and tough leaves with a robust stem (Fig. 1, b), mostly with bigger flowers compared to that of V. planifolia. Its pods are bigger, with lower vanillin content (Grisoni and Nany, Reference Grisoni and Nany2021).
Parentage analysis confirmed V. planifolia and V. pompona as ancestors of Tsitaitra hybrid
The investigation of hybrid types and their parentage confirmed that the hybrid Tsitaitra is a result of the cross between V. planifolia and V. pompona, as evidenced by the identification of shared alleles (Figure S4). Considering the rate of shared alleles (about 90% with V. pompona) with the presumed parents, the analysis further revealed that the Tsitaitra accessions in the collection may represent backcrosses of (V. planifolia X V. pompona) with at least two times V. pompona rather than a simple backcross between (V. planifolia X V. pompona) with V. planifolia as previously reported (Grisoni and Nany, Reference Grisoni and Nany2021). Findings are in agreement with the results obtained from the UPGMA analysis, where Tsitaitra I closely clustered with V. pompona II (Fig. 3), and is consistent with the ADMIXTURE results, which indicated that Tsitaitra I was not clearly separated from V. pompona II at all values of K (Fig. 5).
In contrast, the analysis of Tsivaky, for which the parents are completely unknown, presented complexities that require further exploration and understanding. Discrepancies between the findings from the PCA plot (Fig. 2) and the shared alleles analysis (Figure S4) indicate potential differences in the genetic background of Tsivaky. While the PCA plot suggests a closer relationship with V. planifolia (Fig. 2), the shared alleles analysis indicates a closer association with V. pompona rather than V. planifolia (Figure S4). Several hypotheses can explain these differences. Firstly, the observed discrepancies may be attributed to the genetic background of Tsivaky, which might be more similar to V. pompona despite its apparent proximity to V. planifolia on the PCA plot (Fig. 2). This divergence could result from shared genetic variants or ancestral relationships that are not adequately captured by the first three principal components. However, both UPGMA (Fig. 3) and ADMIXTURE (Fig. 5) confirmed the separation of Tsivaky from V. planifolia. Secondly, the presence of polyploidy (Bory et al., Reference Bory, Catrice, Brown, Leitch, Gigant, Chiroleu, Grisoni, Duval and Besse2008c; Rodolphe et al., Reference Rodolphe, Bory, Grisoni and Besse2011) in Tsivaky could also contribute to the observed differences. Polyploidy, characterized by multiple sets of chromosomes, can introduce complexities in inheritance patterns and allele combinations. In the case of Tsivaky, polyploidy may result in a more intricate genetic makeup, potentially influencing the observed relationships with presumed parents. The presence of multiple copies of certain alleles and the existence of homoeologous chromosomes derived from different ancestral species in polyploids can make it challenging to identify direct allele sharing (Soltis et al., Reference Soltis, Soltis and Tate2004). Additionally, the absence of the parent population within the studied accessions could contribute to the discrepancies observed in the analysis of Tsivaky. If one of the presumed parents is not included in the analysed samples, it becomes challenging to accurately assess the genetic contributions and relationships. The missing parent population hypothesis suggests that the true genetic makeup of Tsivaky may involve a parent species that was not included in the study. This hypothesis finds support in the presence of a high number of private alleles in the Tsivaky genetic group (Table 1), indicating potential inheritance from species not accounted for in the analysis. The inclusion of a larger number of accessions is crucial for obtaining a more accurate understanding of the genetic composition of Tsivaky. These findings emphasize the need for further research and exploration to unravel the underlying genetic factors and obtain a comprehensive understanding of the hybridization processes in vanilla.
BCFtools, a practical tool for vanilla genetic diversity study
Pipelines widely used in population genetic diversity and structure studies include Stacks, BCFtools and GATK were employed in this study to assess their performance in SNP genotyping and population structure analysis in vanilla. The comparison was focused on several parameters, such as the ability of the tools to detect the genetic structure of vanilla accessions, SNP count, missing data rates, loci coverage, heterozygous genotypes and computational efficiency (Figures S2, S3, Table S3).
Our results clearly indicate that Stacks is the least suitable pipeline for studying the genetic structure of vanilla compared to the other two pipelines. The phenotypic groups showed a mixed distribution when using Stacks (Figures S2, S3), suggesting that this tool may not accurately reveal the population structure within the vanilla germplasm. Although Stacks has been successfully used in several studies (Saenz-Agudelo et al., Reference Saenz-Agudelo, DiBattista, Piatek, Gaither, Harrison, Nanninga and Berumen2015; Esposito et al., Reference Esposito, Cardi, Campanelli, Sestili, Díez, Soler, Prohens and Tripodi2020; Feng et al., Reference Feng, Zhao, Li, Zhang, Qu, Li, Li, Lin and Pu2020), it did not perform well in the context of vanilla, as indicated by the present study. Vanilla is known for its high level of heterozygosity (Hu et al., Reference Hu, Resende, Bombarely, Brym, Bassil and Chambers2019), requiring robust and sensitive tools capable of accurately detecting and characterizing this genetic diversity. The success of BCFtools and GATK may be, hence, attributed to their ability to identify more likely heterozygous loci (Li, Reference Li2011; Li and Wren; Reference Li and Wren2014; Table S3). Our research supports the notion that preliminary evaluation of several pipelines for a given dataset is crucial for investigating genetic diversity (Olson et al., Reference Olson, Lund, Colman, Foster, Sahl, Schupp and Zook2015). On the other hand, GATK and BCFtools yielded similar that were consistent with the SNPs commonly detected by all three pipelines (Figures S2, S3) outputs, although, GATK required more computational resources in terms of time and memory than BCFtools (Table S3). The low number of SNPs common to all three pipelines suggests the presence of pipeline-specific biases, indicating that each approach has its own particularities and limitations. However, among the three pipelines, BCFtools, when combined with stringent filtering, emerged as a suitable option for genome-wide genetic investigation in vanilla. It demonstrated reasonable computational requirements while providing satisfactory output quality.
Conclusions
The genetic data obtained from this study provides valuable insights into the genetic diversity and structure of the vanilla population, with implications for taxonomy, breeding and conservation programmes. The genetic clustering analysis clarified the parentage and hybridization processes within the collection, contributing to a better understanding of genetic relationships and the refinement of vanilla taxonomy. The identification of distinct subgroups and the presence of private alleles within phenotypic groups suggests ongoing divergence and the existence of distinct genetic lineages. These findings can guide breeders in selecting appropriate parental lines to enhance specific traits of interest, such as disease resistance, pod characteristics and vanillin content. Moreover, the conservation of Madagascar's genetic resources is crucial, as the study indicates a potential loss of genetic diversity by comparing the genetic resources evaluated by the present study to that of the ancient breeding programme (Grisoni and Nany, Reference Grisoni and Nany2021). The knowledge gained from genetic clustering can guide conservation efforts, prioritize populations for conservation, and inform in-situ or ex-situ conservation strategies. Further research on a larger scale is essential to gather comprehensive information about Madagascar's vanilla gene pool.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S1479262123000631.
Data availability statement
ddRAD sequences for the accessions in this study are deposited at NCBI SRA (BioProject no. PRJNA988271, BioSample accession no SAMN36015065 - SAMN36015109, as listed in Table S1).
Acknowledgements
This research was supported by the Académie de Recherche et d’ Enseignement Supérieur (ARES), Belgium (Programme PRD-PFS 2016 - Madagascar – Vanille). We sincerely acknowledge the support from the Rama Export and Takasago companies that provided us the samples. We thank the following laboratories for their support during our lab work: molecular biology laboratory of the Earth and Life Institute - Agronomy - Université catholique de Louvain – Belgium; biotechnology and microbiology laboratory; Fundamental and Applied Biochemistry Department, Université d’ Antananarivo, Madagascar and the ‘Validation des Nouvelles Pistes et Opportunités’ laboratory, IMRA, Antananarivo, Madagascar.
Competing interests
None.