


Introduction
Dipetalonema gracile is a common filarioid nematode that inhabits the peritoneal cavity of the primate host (Travi et al., Reference Travi, Eberhard and Lowrie1985). In the case of massive parasitism, the host is roughened, malnourished, anorexic and eventually dies, thus the infection poses a serious threat to the health of squirrel monkey (Saimiri sciureus) populations (Notarnicola et al., Reference Notarnicola, Agustin and Gardner2007). In the present era of highly advanced technology, mitochondrial DNA (mtDNA) has been characterized as a simple structure with small molecular mass, high mutation rate, fast evolution, and unique maternal hereditary and rare genetic recombination (Bandyopadhyay et al., Reference Bandyopadhyay, Stevenson and Cady2006; Hu and Gasser, Reference Hu and Gasser2006; Cameron et al., Reference Cameron, Johnson and Whiting2007). Therefore, the use of mtDNA as a genetic marker is more effective in identifying the hidden species and genotypes of parasites (Liu et al., Reference Liu, Wang and Song2013). Lefoulon et al. (Reference Lefoulon, Bain and Bourret2015) analysed the cox1 and 12S rDNA genes of 48 species of nematodes of Onchocercidae, and found that these nematodes were mainly clustered in the genera Dipetalonema, Setaria, Onchocerca, Serofilaria and Dirofilaria. Sazmand et al. (Reference Sazmand, Eigner and Mirzaei2016) amplified the cox1 gene of the microfilariae and adult stages of Dipetalonema evansi from Camelus dromedarius in the south-east of Iran, and found that the cox1 gene could be used for the accurate diagnosis of nematode infection at different stages. With the development of polymerase chain reaction (PCR) and sequencing technologies, many important breakthroughs have been made in studies on the structural characteristic, gene composition and function, and genetic evolution of the mtDNA from parasitic nematodes (Xu et al., Reference Xu, Qiu and Liu2015; Hu et al., Reference Hu2016; Shi et al., Reference Shi2017). However, little is known about the mitochondrial genome of Dipetalonema nematodes.
This study aimed to identify a suspected D. gracile worm from a dead squirrel monkey in a zoo in Guangzhou, China, and to amplify its complete mitochondrial genome sequence by conventional or long-range PCR and sequence analysis.
Materials and methods
Parasites and DNA extraction
Three worms were collected from the abdominal cavity of a dead squirrel monkey in a zoo in Guangzhou in April 2016, fixed in 70% ethanol and stored at −20°C until use. Individual worms were put in centrifuge tubes and flushed three times with double-distilled water (ddH2O). Total genomic DNA from individual worms was extracted using the Wizard® SV Genomic DNA Purification System (Promega, Madison, Wisconsin, USA) according to the manufacturer's instructions and stored at −20°C.
Molecular identification
The primer CX1 (F): 5'-GACCAGGAAGTAGTTGAA-3' and its complementary primer CX1 (R): 5'-CAGCCTCACTAATAATACCA-3' were designed according to published cox1 gene sequences of Dipetalonema nematodes in GenBank (Lefoulon et al., Reference Lefoulon, Bain and Bourret2015). PCR reactions were performed in 25 μl, including 12.5 μl of ExTaq polymerase (TaKaRa, Kusatsu, Shiga, Japan), 0.5 μl of each primer (50 pmol/μl), 2 μl of DNA sample and 9.5 μl of ddH2O. The cycling conditions were initial denaturation at 94°C for 5 minutes, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 52°C for 30 s and extension at 72°C for 1 minute, then a final extension at 72°C for 10 minutes. Amplified fragments were analysed with ethidium bromide stained agarose gel electrophoresis, purified using a DNA gel extraction kit (Omega, Georgia, USA). The purified PCR products were connected with pMD18-T (TaKaRa, Kusatsu, Shiga, Japan) overnight, then transferred into DH5α Competent Cells (TaKaRa, Kusatsu, Shiga, Japan). Positive clones were screened by bacterial PCR and sent to Shanghai Sangon Co., Ltd for sequencing. Homologous comparison was conducted with cox1 gene sequences of Onchocercidae nematodes from the GenBank database. Finally, the cox1 gene sequences of 12 species of Spirurida nematodes were compared using the MEGA6 software, and the best model was selected by ProtTest 2.4. Using Angiostrongylus cantonensis (AB684376) as outgroup, the phylogenetic tree was constructed by maximum likelihood (ML) and maximum parsimony (MP) methods (Zhan et al., Reference Zhan, Li and Xiao2001).
PCR amplification of complete mitochondrial genome
According to the complete mitochondrial genome sequence of Dirofilaria immitis (NC005305) published in GenBank, seven pairs of primers (table 1) were designed in their conserved regions to amplify the entire mitochondrial genome sequence of D. gracile. These primers were synthesized by Shanghai Sangon Company in China. PCR reactions for a ≤ 2 kb fragment were performed in 50 μl, including 25 μl of Premix PrimeStar Max (TaKaRa, Kusatsu, Shiga, Japan), 1 μl of each primer (50 pmol/μl), 4 μl of DNA samples and 19 μl of ddH2O. PCR conditions used were initial denaturation at 94°C for 5 minutes, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 52°C for 30 s and extension at 72°C for 1 minute, followed by a final extension at 72°C for 10 minutes. Long PCR reactions for a > 2 kb fragment were performed in 50 μl, including 25 μl of Premix PrimeStar Max, 1 μl of each primer (50 pmol/μl), 4 μl of DNA samples and 19 μl of ddH2O. The cycling conditions were initial denaturation at 94°C for 5 minutes; followed by denaturation at 94°C for 30 s, annealing at 42–53°C for 30 s and extension at 68°C for 1.5 minutes for 10 cycles; followed by initial denaturation at 94°C for 5 minutes; denaturation at 94°C for 30 s, annealing at 50–58°C for 30 s and extension at 72°C for 1.5–2.0 minutes for 25 cycles; and then a final extension at 72°C for 7 minutes. Amplified PCR products were analysed with ethidium bromide stained agarose gel electrophoresis, purified using a DNA gel extraction kit (Omega, Georgia, USA). The purified PCR products were connected with pMD18-T (TaKaRa, Kusatsu, Shiga, Japan) overnight, then transferred into DH5α Competent Cells (TaKaRa, Kusatsu, Shiga, Japan). Positive clones were screened by bacterial PCR and sent to Shanghai Sangon Co., Ltd for sequencing.
Table 1. Primers used for PCR amplification of the Dipetalonema gracile mitochondrial genome.

Sequence analysis
The high-quality sequences obtained using BioEdit version 7.0 were assembled by seqMan software within DNAStar 5.0 (Tamura et al., Reference Tamura, Peterson and Peterson2011) and adjusted manually. Online software (http://dogma.ccbb.utexas.edu/) was combined with MegAlign software in DNAStar 5.0 (Tamura et al., Reference Tamura, Peterson and Peterson2011) to identify gene boundaries and composition, as well as translation initiation and termination codons. The AT contents were calculated using Editseq software in DNAStar 5.0 (Tamura et al., Reference Tamura, Peterson and Peterson2011). The 22 tRNA genes were identified with the aid of the tRNA scan program, available at http://lowelab.ucsc.edu/tRNAscan-SE/, combined with artificial proofreading using Dirofilaria immitis. The rRNA genes were identified by aligning sequence with those of D. immitis (Hu et al., Reference Hu2003). Their secondary structures were predicted by comparing them with the published structures of D. immitis (Hu et al., Reference Hu2003).
Results
Molecular identification of D. gracile
The amplified fragment of the cox1 gene was approximately 600 bp in length, which is consistent with the expected size. The sequencing results showed that the cox1 gene was 632 bp long. BLAST analysis indicated highest similarity (98.90%) with D. gracile (KP760182). Phylogenetic analyses showed that the attained sequence clustered in the same branch as Dipetalonema gracile (KP760181) (fig. 1). Thus, the suspected worm was identified as D. gracile.
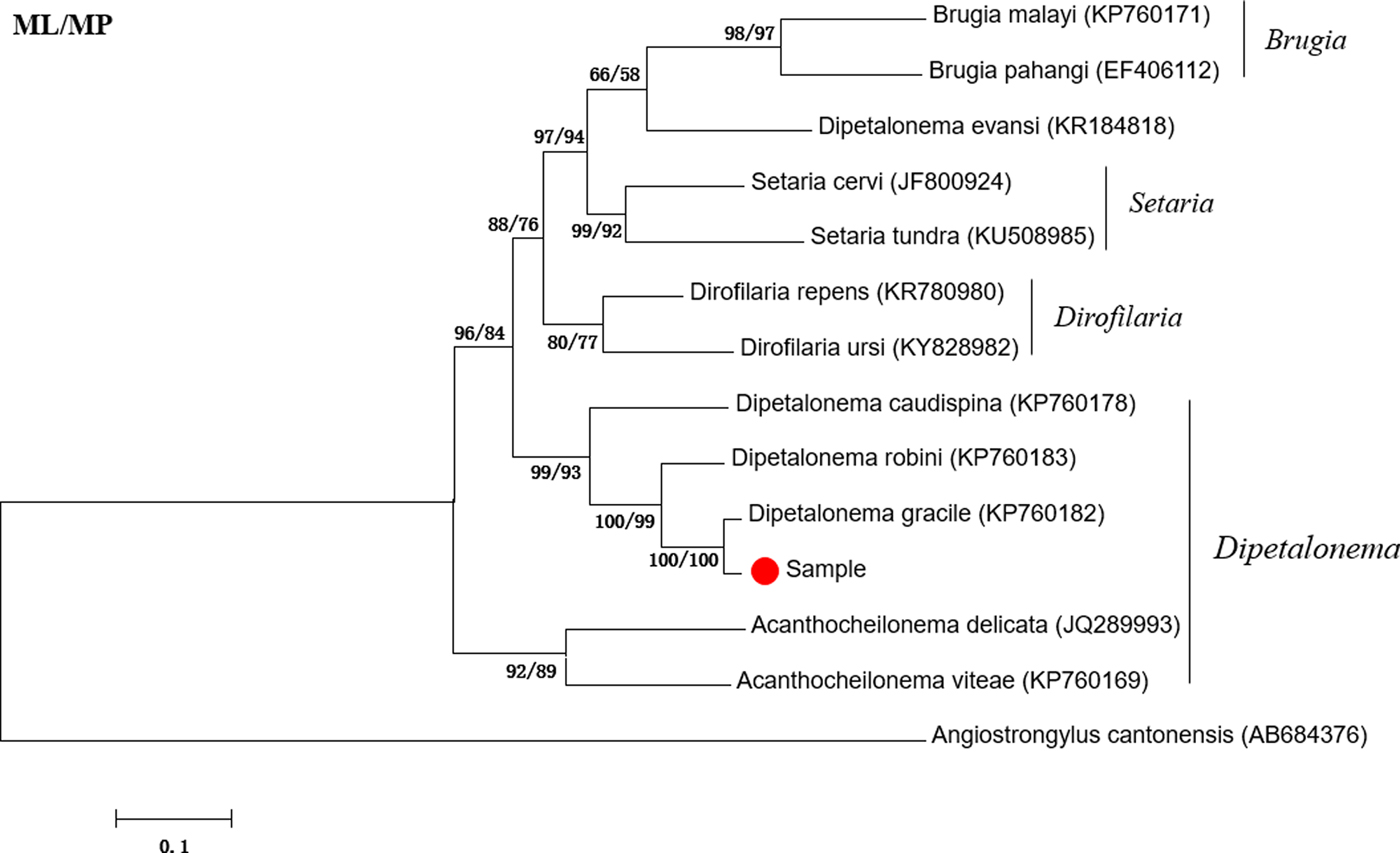
Fig. 1. Phylogenetic tree based on the cox1 gene of Dipetalonema gracile and other Onchocercidae nematodes by maximum likelihood (ML) and maximum parsimony (MP) methods.
Amplification of complete mitochondrial genome
The amplified fragments from seven pairs of primers (F1–F7) for the complete mitochondrial genome of D. gracile were 900 bp, 1003 bp, 1026 bp, 3179 bp, 2093 bp, 3212 bp, and 2068 bp in size, respectively, which are consistent with the expected fragments, without non-specific bands.
General features of D. gracile mitochondrial genome
The entire mitochondrial genome sequence of D. gracile was 13,584 bp in length. There were 36 genes, including 22 tRNA genes, 12 protein-coding genes, two rRNA genes, one AT-rich region and one small non-coding region (SNCR), which constituted a closed circular structure (fig. 2). There were 16 intergenic regions, ranging from 1 to 9 bp (table 2). The nucleotide composition was A = 16.89%, G = 20.19%, T = 56.22%, and C = 6.70%. Therefore, A + T = 73.11%, with obvious AT preference.
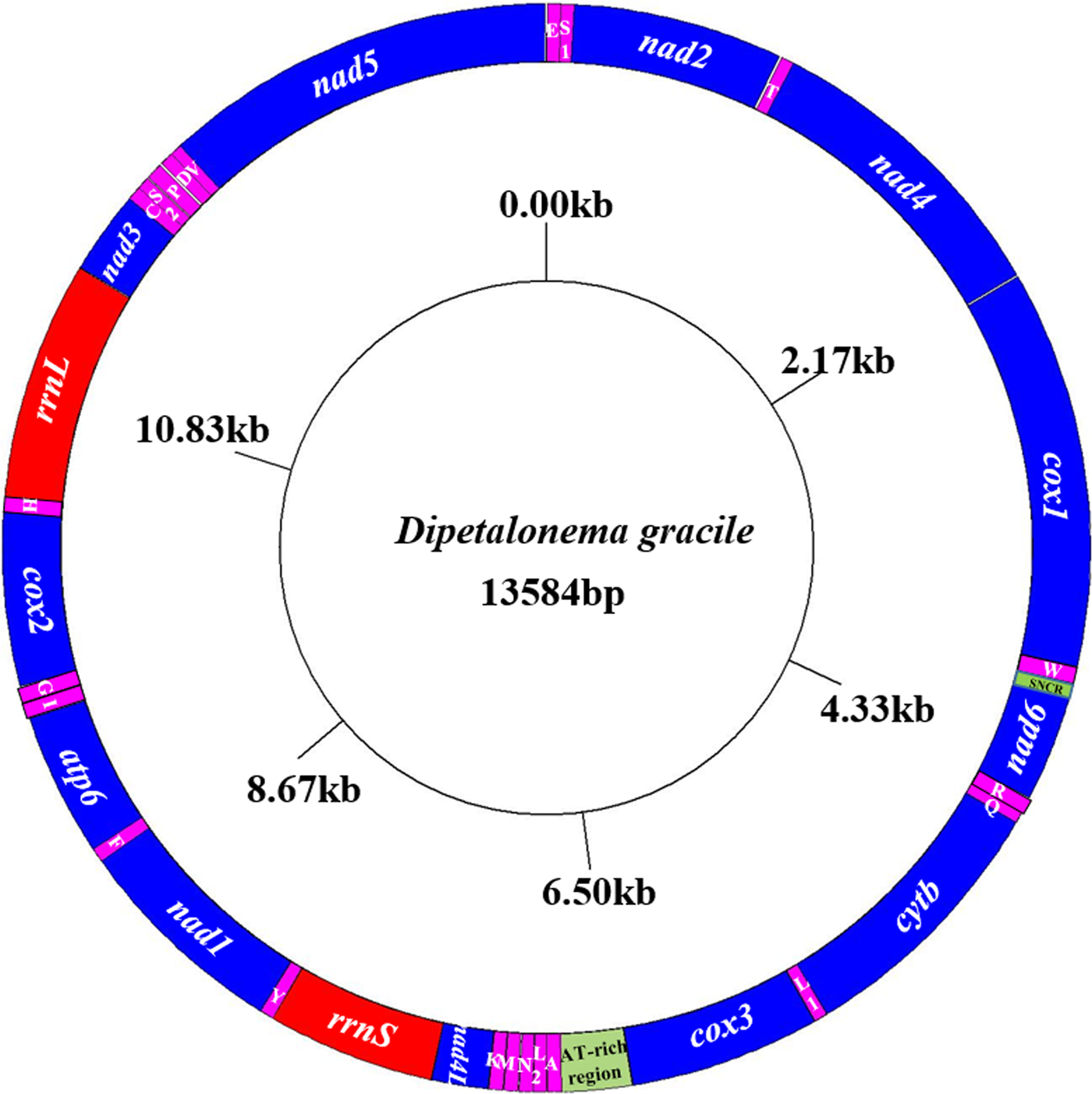
Fig. 2. Arrangement of the mitochondrial genome of D. gracile. All genes are predicted to be transcribed in a clockwise direction, and the tRNA genes are designated by single-letter abbreviations for the corresponding amino acids.
Table 2. Organization of the D. gracile mitochondrial genome.

Protein-coding genes
The lengths of 12 protein-coding genes of D. gracile were stable. Except nad4 and cox1, all other protein-coding genes were separated by tRNA genes (table 2). The 12 protein-coding genes were biased towards A and T, where the lowest gene in AT content was cox1 (67.06%) and the highest was nad6 (77.65%) (table 3). They used TAT, TTG, GTA, CTT, GTT and ATT as the start codons. Among them, TTG was the most common (50.00%), followed by ATT (16.67%) and the others (8.30%). The use of the termination codons was more variable; there were complete TAG (41.67%), TAA (25.00%) and TTA (8.30%) codons, and incomplete T (50.00%) stop codons.
Table 3. Nucleotide composition (%) of 12 protein-coding genes of the D. gracile mitochondrial genome.

Transfer RNA genes
The 22 tRNA genes in the mitochondrial genome of D. gracile formed a local double-helix structure by base pairing. The acceptor arm on the top was composed of 7 base-pairs, and the anticodon area included a stem of 5 base-pairs and a loop of 7 bases. These structures folded to form a stable and atypical cloverleaf pattern 52–61 bp long. The trnS1AGN and trnS2UCN lacked D-loop, where 4 or 8 bases were connected to amino acid acceptor arm and anticodon loop, and 6 or 5 bases and 3 base-pairs together made up the TΨC loop. The remaining 20 tRNA genes lacked TΨC loops, where 5 to 8 bases were connected to amino acid acceptor arm and anticodon loop, and 4 to 11 bases and 4 base-pairs together made up the TV-loop (fig. 3).

Fig. 3. Secondary structures predicted for the 22 tRNA genes in the mitochondrial genome of D. gracile. Canonical base pairs C:G and U:A are indicated by dashes, and G:U pairs by dots.
Ribosomal RNA genes
The two rRNA genes in the mitochondrial genome of D. gracile encoded a large subunit 16S (rrnL) and small subunit 12S (rrnS). They were located between trnH and nad3, and trnY and nad4L, respectively. The lengths of rrnL and rrnS genes were 968 bp and 685 bp, respectively. The AT contents were 72.62% and 75.04%, respectively. Two rRNA genes were single-stranded and relatively conservative in secondary structures. Through A-U and G-C pairings, and even A-A, U-U, G-U and A-G unstable pairings, they formed multiple stem-loop structures (fig. 4).

Fig. 4. Predicted secondary structure of the mitochondrial rrnL (a) and rrnS (b) inferred for D. gracile. Canonical base pairs C:G and U:A are indicated by dashes, G:U pairs by large dots, other non-canonical pairings by small dots, and proposed tertiary interactions by lines.
AT-rich and small non-coding region
The AT-rich region in the mitochondrial genome of D. gracile was located between trnA and nad3, without gene interval. The sequence length was 286 bp, and AT content was up to 80.77%, higher than the 12 protein-coding genes. The small non-coding region (SNCR) was 46 bp in length, and was located between trnW and nad6, without stem-loop structure.
Discussion
At present, there are only a few morphological and developmental descriptions of D. gracile (Travi et al., Reference Travi, Eberhard and Lowrie1985). In this study, molecular identification of a suspected D. gracile worm from a squirrel monkey was conducted, confirming that the worm was D. gracile, and its complete mitochondrial genome sequence was obtained for the first time. The mitochondrial genome of D. gracile was 13,584 bp in length, and its structure and nucleotide composition are basically similar to other nematodes of Secernentea. This may be because the mitochondrial genes could be under similar evolutionary pressures during the genetic process (Hyman and Azevedo, Reference Hyman and Azevedo1996; Gao et al., Reference Gao, Qiu and Zhang2017). Moreover, mitochondrial dysfunction may result from changes in the structures and lengths of mitochondrial genes. The stop codons of D. gracile protein-coding genes were TAG and TAA, which is consistent with most nematodes (Okimoto et al., Reference Okimoto, Macfarlane and Wolstenholme1990). The use of incomplete codon T also occurred, possibly as a result of post-transcriptional processing when AA is inserted after T to act as the stop codon for protein translation (Ojala et al., Reference Ojala, Montoya and Attardi1981). Among the 12 protein-coding genes, the AT content of nad6 gene was the highest (77.65%), and that of cox1 gene was the lowest (67.06%). Therefore, it is speculated that the cox1 gene may be under relatively high selection pressure (Dingley et al., Reference Dingley, Polyak and Ostrovsky2014), while the nad6 gene may be under relatively low selection pressure. As a whole, the 12 protein-coding genes had obvious AT preference; a higher AT preference makes the gene structure more stable and may reduce the probability of gene mutation. This makes multiple protein-coding genes in mitochondria ideal molecular markers for studying molecular classification, phylogenetic evolution, and population genetic variation of the parasite.
In the present study the 22 tRNA genes of D. gracile formed a local double-helix structure, as in most nematodes. With the exception of trnS1AGN and trnS2UCN, the remaining 20 tRNA genes lacked a TΨC loop, showing a TV-loop structure. The polymorphism of the tRNA gene structure may suggest that there are metabolic pathways in this nematode that are different from other organisms (Zhang and Kong, Reference Zhang and Kong1997). The rRNA gene of D. gracile had multiple unstable pairings, forming many stem and loop structures of different sizes. Such secondary structures were complex but relatively conserved. This means that the differences in rRNA genes among related species can be applied to the classification and phylogenetic studies of nematodes. In addition, there were many common mismatches in the secondary structure of the tRNA and rRNA genes of D. gracile. However, no mechanism has been found to correct mitochondrial gene mismatch (Pont-Kingdon et al., Reference Pont-Kingdon, Vassort and Warrior2000). The PCR amplification of the non-coding region of D. gracile was difficult, possibly because it does not participate in mitochondrial transcription, with relatively low evolutionary pressure and high mutation rate (Blouin, Reference Blouin2002). It is worth noting that the base composition of the D. gracile mitochondrial genome had obvious AT bias, which may increase the mutation rate of nucleic acids and the substitution rate of amino acids, making the silent sites more rapidly saturated. This evolutionary trend is conducive to the study of genetic polymorphism and phylogenetics (Zhang et al., Reference Zhang2015).
In conclusion, this study identified D. gracile from an infected squirrel monkey in China and obtained its complete mitochondrial genome sequence for the first time, thus enriching the mitochondrial gene database of Dipetalonema nematodes. It lays a foundation for studying the classification and genetic evolutionary relationships of Dipetalonema nematodes.
Financial support
This work was funded by the National Natural Science Foundation of China (Grant no. 31672541) and the Science and Technology Planning Project of Guangdong Province, China (Grant no. 2014A020214005).
Conflict of interest
None.







