


So far many countries have developed national clinical guidelines for the treatment of major depressive disorder (Depression Guideline Panel, 1993a , b ; Reference Rush, Crismon and TopracRush et al, 1998; Reference Mulrow, Williams and TrivediMulrow et al, 1999; American Psychiatric Association, 2000; Reference Anderson, Nutt and DeakinAnderson et al, 2000; Reference Kennedy, Lam and CohenKennedy et al, 2001; National Institute for Clinical Excellence, 2004). In these guidelines pharmacotherapy is among the most important treatments, and in many countries selective serotonin reuptake inhibitors (SSRIs) have become the first-line antidepressants. It is less clear what should be done in those 40–50% of patients who do not respond to the first antidepressant administered (Reference Thase, Rush, Bloom and KupferThase & Rush, 1995; Reference Kroenke, West and SwindleKroenke et al, 2001). Strategies in case of non-response have been published in several narrative reviews (Reference Thase and RushThase & Rush, 1997; Reference NelsonNelson, 1998; Reference Crismon, Trivedi and PigottCrismon et al, 1999; Fava, Reference Fava2000a ,Reference Fava b ; Reference O'Reardon, Brunswick and AmsterdamO'Reardon et al, 2000; Reference MarangellMarangell, 2001; Reference Trivedi and KleiberTrivedi & Kleiber, 2001; Reference Hirschfeld, Montgomery and AgugliaHirschfeld et al, 2002; Reference Kennedy, McIntyre and FalluKennedy et al, 2002; Reference AndersonAnderson, 2003; Reference Kennedy and McDonoughKennedy & McDonough, 2003; Reference McIntyre, Muller and ManciniMcIntyre et al, 2003; Reference NelsonNelson, 2003) and in one systematic review (Reference Stimpson, Agrawal and LewisStimpson et al, 2002). Three major strategies for non-response are recommended: dose escalation, augmenting the antidepressant by adding a second drug, and switching to another antidepressant of the same or a different class.
Available dose-finding studies do not provide evidence for initiating pharmacotherapy for major depressive disorder with SSRIs in higher than standard doses (Reference Altamura, Montgomery and WernickeAltamura et al, 1988; Reference Beasley, Bosomworth and WernickeBeasley et al, 1990; Reference Dunner and DunbarDunner & Dunbar, 1992; Reference Tignol, Stoker and DunbarTignol et al, 1992; Reference Montgomery, Pedersen and TanghojMontgomery et al, 1994). For non-responders, all guidelines recommend dose escalation as the appropriate strategy, instead of continuing an apparently inadequate regimen (Depression Guideline Panel, 1993a ; b ; Reference Rush, Crismon and TopracRush et al, 1998; Reference Mulrow, Williams and TrivediMulrow et al, 1999; American Psychiatric Association, 2000; Reference Anderson, Nutt and DeakinAnderson et al, 2000; Reference Kennedy, Lam and CohenKennedy et al, 2001). Only the National Institute for Clinical Excellence (NICE) guideline is less definite (National Institute for Clinical Excellence, 2004), advising that if ‘there are no significant side-effects, a gradual increase in dose should be considered’. Moreover, surprisingly little systematic evidence is provided to support these recommendations. Because of the above recommendations and because of its simplicity, dose escalation is widely practised and often the first strategy applied (Reference Byrne and RothschildByrne & Rothschild, 1997; Reference Shergill and KatonaShergill & Katona, 1997; Reference Fredman, Fava and KienkeFredman et al, 2000; Reference Mischoulon, Nierenberg and KizilbashMischoulon et al, 2000). The aim of our study was to systematically review the evidence for dose escalation of SSRIs in major depressive disorder.
METHOD
Design of studies to be included
Ideally the design of dose-escalation studies is randomisation of non-responders to higher doses of an antidepressant or placebo after some weeks of a standard-dose regimen. In this review we consider three other methodological requirements for such studies. First, dose escalation should be deferred to 3–6 weeks after initiation of treatment, because several weeks are required for antidepressants to have clinical effect (Reference MischoulonMischoulon, 1997). The practice of dose escalation and the demonstration of a dose–response relationship is based on selection of ‘true’ nonresponders (Reference Baker and WoodsBaker & Woods, 2003). As this might take 6–10 weeks (Reference Quitkin, Petkova and McGrathQuitkin et al, 2003), dose-escalation studies with early randomisation diminish the possibility of proving the usefulness of dose escalation. The inclusion of unidentified late responders in both arms of the study reduces the contrast between the intervention and control. Second, an outstanding study will have sufficient power to be able to demonstrate a clinically relevant difference (e.g. 20%) between treatment arms and, third, will describe the method of dose escalation and describe the early drop-out rates because of dose escalation.
Identification and selection of articles
First, systematic literature searches (updated 10 February 2005) were performed in four databases (MEDLINE, EMBASE, CINAHL, PsycInfo; all indexed years). As there are no specific keywords for dose-escalation studies, sensitive searches were performed with the following terms: (((dose[textword(tw)] OR dosage[tw]) AND increase[tw]) OR ((dose[tw] OR dosage[tw]) AND maxim*[tw]) OR (upward[tw] AND titrat*[tw])) OR dose–response relationship, drug[MeSH], in combination with the Cochrane Collaboration search-filter for randomised controlled trials and systematic reviews, the Cochrane Collaboration Depression Anxiety and Neurosis group search-filter for major depressive disorder and MeSH-terms and text words for SSRIs. Primary selection (independently by H.R. and J.H.) was based on design and focused on dose–response relationships for SSRIs, by screening title and abstract of the article. Agreement on exclusion of irrelevant articles was 99.1%, with Cohen's kappa for interrater agreement 0.62 (which is a substantial agreement (Reference Munoz and BangdiwalaMunoz & Bangdiwala, 1997)). Discrepancies between initial selection were resolved by discussion and consensus.
Second, all potentially relevant articles were judged according to specific inclusion and exclusion criteria (criteria available from H.R. on request). In case of doubt, an article was read fully and assigned afterwards. Additionally, relevant cross-references were retrieved. Double publications were considered together to reveal the maximum available information.
Critical appraisal and summary
Next, selected articles were critically appraised and abstracted by H.R., using standardised forms derived from the Dutch Institute of Healthcare Improvement (Kwaliteitsinstituut voor de Gezondheidszorg CBO, 2000) and the Agency for Healthcare Policy and Research (Reference Mulrow, Williams and TrivediMulrow et al, 1999). The items used for critical appraisal were the same as proposed by the Scottish Intercollegiate Guideline Network (2001) and Sackett et al (Reference Sackett, Straus and Richardson2000). Each study was assigned a ‘level of evidence’ (Table 1). Levels of evidence were based on the methodological robustness of studies. For the results, the highest level of evidence of the supporting scientific evidence (A1–D) was used.
Table 1 Levels of evidence in therapeutic studies

| Level | Type of study |
|---|---|
| A1 | Systematic review including at least some studies of A2 level. Consistent results (homogeneity) across the included trials |
| A2 | Randomised controlled (double-blind) trial of good methodological quality, adequate size and consistency of results |
| B | Randomised clinical trial of lower methodological quality or inadequate size. Other comparative research (e.g. non-randomised trial, comparative cohort study, case—control study) |
| C | Uncontrolled, open study |
| D | Expert opinion, e.g. guideline panel members |
To assess judgement bias of the person who performed the critical appraisal, inter-rater variation was determined in a slightly different set of 12 publications. We all critically appraised four publications, and agreement for the appraisal items was expressed by Cohen's kappa. Kappa values were 0.49 (for validity of the study), 0.86 (for concealment of allocation); complete agreement existed for randomisation of the study, level of evidence and data extraction (kappa=1.0). This is in line with other reports of interrater agreement in appraisal of psychiatric research (Reference Moncrieff, Churchill and DrummondMoncrieff et al, 2001).
A qualitative summary with discussion of the results, restrictions, methodological flaws and external validity of the studies was described in an evidence table and a separate document, of which a summary is provided in this paper. Because of the apparent heterogeneity in timing of the dose escalation between the studies, results were not pooled in a meta-analysis.
RESULTS
Search results and selection of studies are presented in Fig. 1. The 11 studies selected for this review are summarised in Table 2. A table of excluded studies is available from H.R. on request.
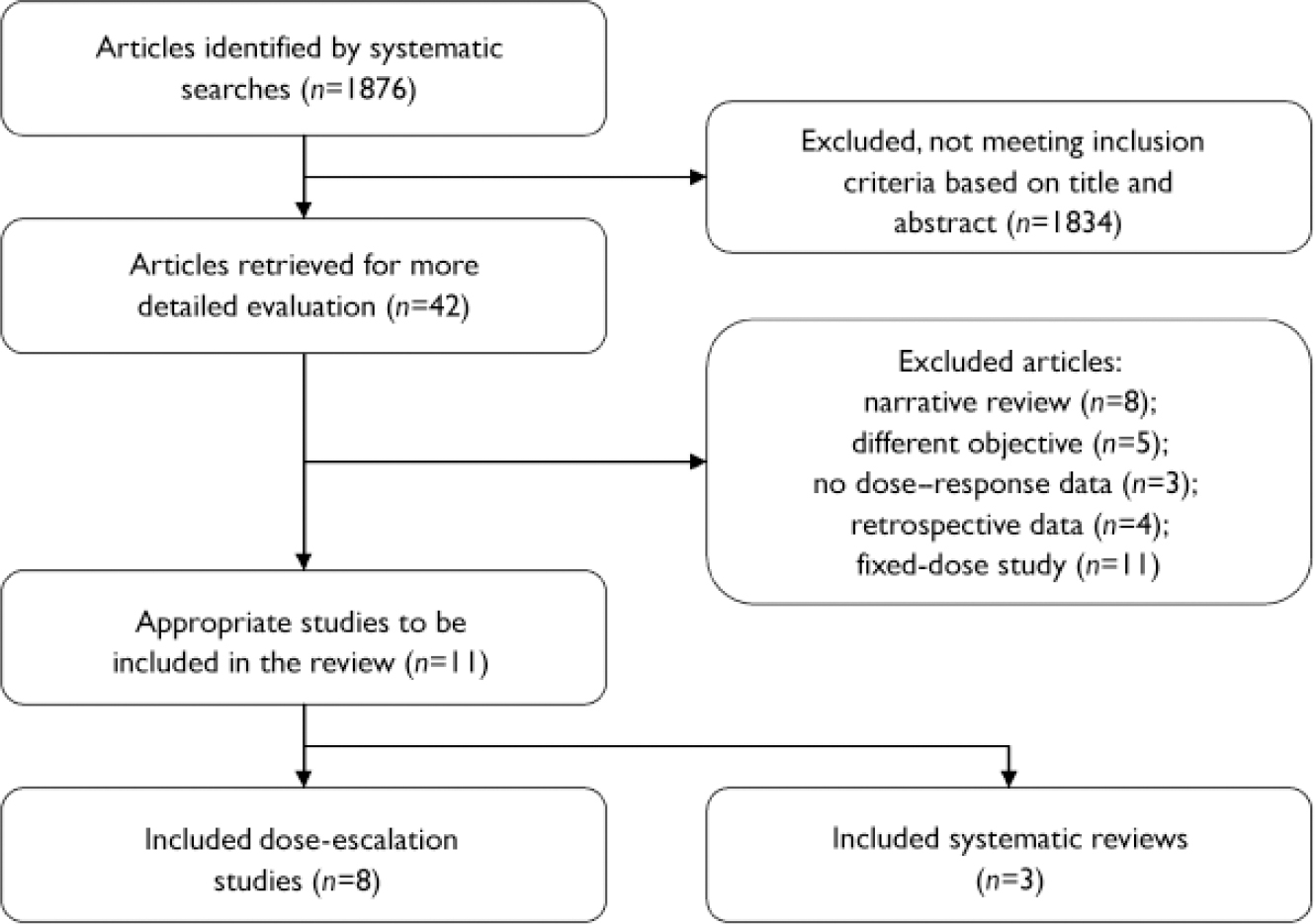
Fig. 1 Selection process for reported studies.
Table 2 Effectiveness of increasing the dose: selected studies
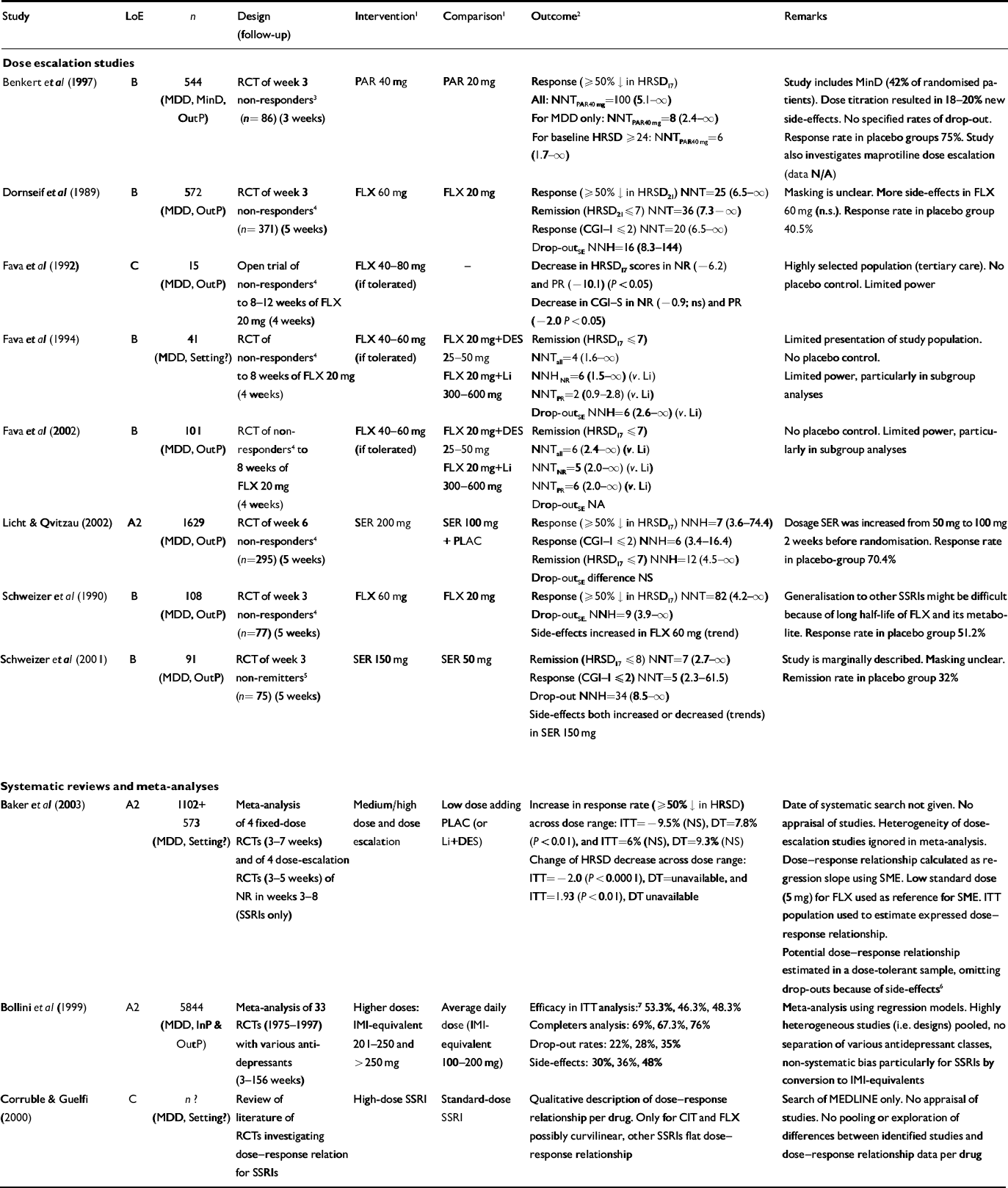
| Study | LoE | n | Design (follow-up) | Intervention1 | Comparison1 | Outcome2 | Remarks |
|---|---|---|---|---|---|---|---|
| Dose escalation studies | |||||||
| Benkert et al (Reference Benkert, Szegedi and Wetzel1997) | B | 544 (MDD, MinD, OutP) | RCT of week 3 non-responders3 (n= 86) (3 weeks) | PAR 40 mg | PAR 20 mg | Response (≥50% ↓ in HRSD17) All: NNTPAR40mg=100 (5.1-∞) For MDD only: NNTPAR40mg=8 (2.4-∞) For baseline HRSD ≥24: NNTPAR40mg=6 (1.7-∞) | Study includes MinD (42% of randomised patients). Dose titration resulted in 18-20% new side-effects. No specified rates of drop-out. Response rate in placebo groups 75%. Study also investigates maprotiline dose escalation (data N/A) |
| Dornseif et al (Reference Dornseif, Dunlop and Potvin1989) | B | 572 (MDD, OutP) | RCT of week 3 non-responders4 (n= 371) (5 weeks) | FLX 60 mg | FLX 20 mg | Response (≥50% ↓ in HRSD21) NNT=25 (6.5-∞) Remission (HRSD21≤7) NNT=36 (7.3-∞) Response (CGI-1 ≤2) NNT=20 (6.5-∞) Drop-outSE NNH=16 (8.3-144) | Masking is unclear. More side-effects in FLX 60 mg (n.s.). Response rate in placebo group 40.5% |
| Fava et al (Reference Fava, Cohen and Rosenbaum1992) | C | 15 (MDD, OutP) | Open trial of non-responders4 to 8-12 weeks of FLX 20 mg (4 weeks) | FLX 40-80 mg (if tolerated) | - | Decrease in HRSD17 scores in NR (-6.2) and PR(-10.1) (P<0.05) Decrease in CGI—S in NR (-0.9; ns) and PR (-2.0 P<0.05) | Highly selected population (tertiary care). No placebo control. Limited power |
| Fava et al (Reference Fava, Rosenbaum and McGrath1994) | B | 41 (MDD, Setting?) | RCT of non-responders4 to 8 weeks of FLX 20 mg (4 weeks) | FLX 40-60 mg (if tolerated) | FLX 20 mg+DES 25-50 mg FLX 20 mg+Li 300-600 mg | Remission (HRSD17 ≤7) NNTall=4 (1.6-∞) NNHNR=6 (1.5-∞) (v. Li) NNTPR=2 (0.9-2.8) (v. Li) Drop-outSE NNH=6 (2.6-∞) (v. Li) | Limited presentation of study population. No placebo control. Limited power, particularly in subgroup analyses |
| Fava et al (Reference Fava, Alpert and Nierenberg2002) | B | 101 (MDD, OutP) | RCT of non-responders4 to 8 weeks of FLX 20 mg (4 weeks) | FLX 40-60 mg (if tolerated) | FLX 20 mg+DES 25-50 mg FLX 20 mg+Li 300-600 mg | Remission (HRSD17 ≤7) NNTall=6 (2.4-∞) (v. Li) NNTNR=5 (2.0-∞) (v. Li) NNTPR=6 (2.0-∞) (v. Li) Drop-outSE NA | No placebo control. Limited power, particularly in subgroup analyses |
| Licht & Qvitzau (Reference Licht and Qvitzau2002) | A2 | 1629 (MDD, OutP) | RCT of week 6 non-responders4 (n=295) (5 weeks) | SER 200 mg | SER 100 mg + PLAC | Response (≥50% ↓ in HRSD17) NNH=7 (3.6-74.4) Response (CGI—1 ≤2) NNH=6 (3.4-16.4) Remission (HRSD17 ≤7) NNH=12 (4.5-∞) Drop-outSE difference NS | Dosage SER was increased from 50 mg to 100 mg 2 weeks before randomisation. Response rate in placebo-group 70.4% |
| Schweizer et al (Reference Schweizer, Rickels and Amsterdam1990) | B | 108 (MDD, OutP) | RCT of week 3 non-responders4 (n=77) (5 weeks) | FLX 60 mg | FLX 20 mg | Response (≥50% ↓ in HRSD17) NNT=82 (4.2-∞) Drop-outSE NNH=9 (3.9-∞) Side-effects increased in FLX 60 mg (trend) | Generalisation to other SSRIs might be difficult because of long half-life of FLX and its metabolite. Response rate in placebo group 51.2% |
| Schweizer et al (Reference Schweizer, Rynn and Mandos2001) | B | 91 (MDD, OutP) | RCT of week 3 non-remitters5 (n=75) (5 weeks) | SER 150 mg | SER 50 mg | Remission (HRSD17 ≤8) NNT=7 (2.7-∞) Response (CGI-I ≤2) NNT=5 (2.3-61.5) Drop-out NNH=34 (8.5-∞) Side-effects both increased or decreased (trends) in SER 150 mg | Study is marginally described. Masking unclear. Remission rate in placebo group 32% |
| Systematic reviews and meta-analyses | |||||||
| Baker et al (Reference Baker, Tweedie and Duval2003) | A2 | 1102+ 573 (MDD, Setting?) | Meta-analysis of 4 fixed-dose RCTs (3-7 weeks) and of 4 dose-escalation RCTs (3-5 weeks) of NR in weeks 3-8 (SSRIs only) | Medium/high dose and dose escalation | Low dose adding PLAC (or Li+DES) | Increase in response rate (≥50% ↓ in HRSD) across dose range: ITT=-9.5% (NS), DT=7.8% (P<0.01), and ITT=6% (NS), DT=9.3% (NS) Change of HRSD decrease across dose range: ITT=-2.0 (P<0.0001), DT=unavailable, and ITT=1.93 (P<0.01), DT unavailable | Date of systematic search not given. No appraisal of studies. Heterogeneity of dose-escalation studies ignored in meta-analysis. Dose—response relationship calculated as regression slope using SME. Low standard dose (5 mg) for FLX used as reference for SME. ITT population used to estimate expressed dose—response relationship. Potential dose—response relationship estimated in a dose-tolerant sample, omitting drop-outs because of side-effects6 Meta-analysis using regression models. Highly heterogeneous studies (i.e. designs) pooled, no separation of various antidepressant classes, non-systematic bias particularly for SSRIs by conversion to IMI-equivalents Search of MEDLINE only. No appraisal of studies. No pooling or exploration of differences between identified studies and dose—response relationship data per drug |
| Bollini et al (Reference Bollini, Pampallona and Tibaldi1999) | A2 | 5844 (MDD, InP & OutP) | Meta-analysis of 33 RCTs (1975-1997) with various antidepressants (3-156 weeks) | Higher doses: IMI-equivalent 201-250 and >250 mg | Average daily dose (IMI-equivalent 100-200 mg) | Efficacy in ITT analysis:7 53.3%, 46.3%, 48.3% Completers analysis: 69%, 67.3%, 76% Drop-out rates: 22%, 28%, 35% Side-effects: 30%, 36%, 48% | |
| Corruble & Guelfi (Reference Corruble and Guelfi2000) | C | n? (MDD, Setting?) | Review of literature of RCTs investigating dose—response relation for SSRIs | High-dose SSRI | Standard-dose SSRI | Qualitative description of dose—response relationship per drug. Only for CIT and FLX possibly curvilinear, other SSRIs flat dose—response relationship |
Characteristics of the studies
Our searches identified eight dose-escalation studies that increased dosages after at least 3 weeks of standard dosage (Reference Dornseif, Dunlop and PotvinDornseif et al, 1989; Schweizer et al, Reference Schweizer, Rickels and Amsterdam1990, Reference Schweizer, Rynn and Mandos2001; Fava et al, Reference Fava, Cohen and Rosenbaum1992, Reference Fava, Rosenbaum and McGrath1994, Reference Fava, Alpert and Nierenberg2002; Reference Benkert, Szegedi and WetzelBenkert et al, 1997; Reference Licht and QvitzauLicht & Qvitzau, 2002). We further found three systematic reviews about dose–response relationships, which included, respectively, three (Reference Bollini, Pampallona and TibaldiBollini et al, 1999), three (Reference Corruble and GuelfiCorruble & Guelfi, 2000) and four (Reference Baker, Tweedie and DuvalBaker et al, 2003) of the eight identified dose-escalation studies.
Across the studies different outcome definitions for end-points were used. In seven articles, response was defined as a reduction of ≥50% in the Hamilton Rating Scale for Depression (HRSD) score (Reference Dornseif, Dunlop and PotvinDornseif et al, 1989; Reference Schweizer, Rickels and AmsterdamSchweizer et al, 1990; Reference Benkert, Szegedi and WetzelBenkert et al, 1997; Reference Licht and QvitzauLicht & Qvitzau, 2002; Reference Baker, Tweedie and DuvalBaker et al, 2003). A Clinical Global Impression (CGI) improvement or severity score ≤2 was used for response in one study (Reference Schweizer, Rynn and MandosSchweizer et al, 2001). Partial response was used in three studies and defined as 25–50% decrease in HRSD score (Fava et al, Reference Fava, Cohen and Rosenbaum1992, Reference Fava, Rosenbaum and McGrath1994, Reference Fava, Alpert and Nierenberg2002). In seven studies, remission-rates were reported. These were defined as HRSD score ≤7 (Fava et al, Reference Fava, Rosenbaum and McGrath1994, Reference Fava, Alpert and Nierenberg2002; Reference Licht and QvitzauLicht & Qvitzau, 2002) or HRSD score ≤8 (Reference Schweizer, Rynn and MandosSchweizer et al, 2001).
Different criteria were applied to decide whether a patient should be randomised: non-response according to CGI (Reference Benkert, Szegedi and WetzelBenkert et al, 1997), <50% decrease in HRSD score (Reference Dornseif, Dunlop and PotvinDornseif et al, 1989; Reference Schweizer, Rickels and AmsterdamSchweizer et al, 1990; Fava et al, Reference Fava, Rosenbaum and McGrath1994, Reference Fava, Alpert and Nierenberg2002) or no remission (HRSD score ≤8 (Reference Schweizer, Rynn and MandosSchweizer et al, 2001)). In the present studies no genetic information of the cytochrome P450 (CYP) system nor drug blood levels were reported.
The three previous reviews all had some methodological problems: Bollini et al (Reference Bollini, Pampallona and Tibaldi1999) pooled studies with completely different designs and drug classes, and applied a dose equivalence strategy that put differential doses of SSRIs together. Baker et al (Reference Baker, Tweedie and Duval2003) also pooled heterogeneous studies with different moments of dose escalation, and used an unusually low reference dose of fluoxetine (5 mg). Corruble & Guelfi (Reference Corruble and Guelfi2000) did not use an adequate search strategy and only described the dose–response relationships found in their identified studies as flat, curvilinear or linear.
We will briefly outline the dose-escalation studies. Dorseif et al (1989) first investigated week-3 non-responders (n=371 out-patients) to fluoxetine, who were randomised to continuation with 20 mg or increase to 60 mg/day for 5 weeks. Response rates were 40.5% and 44.7%, respectively, and remission rates 33.3% and 36.2%, respectively. Drop-out rates because of side-effects were significantly different at 5.3% and 11.6%, respectively. Schweizer et al (Reference Schweizer, Rickels and Amsterdam1990) investigated 77 non-responsive out-patients after 3 weeks’ administration of fluoxetine (20 mg/day), with a randomisation to placebo increase or dose escalation up to 60 mg/day for 5 weeks. Response rates were 51.2% and 50%, respectively, with non-significant drop-out rates of 4.9% v. 16.7%. In a similar study, Schweizer et al (Reference Schweizer, Rynn and Mandos2001) studied dose escalation of sertraline in out-patient non-remitters after 3 weeks of sertraline (50 mg/day, n=75). Doses were randomly either kept at 50 mg/day or increased to 150 mg/day. Remission rates after 5 weeks were 32% and 47%, respectively (non-significant). Specified drop-out rates because of side-effects were not reported.
Fava et al (Reference Fava, Cohen and Rosenbaum1992) first openly treated 15 out-patients (who were week-8 non-responders to fluoxetine at 20 mg/day) with increased doses of fluoxetine titrated up to 80 mg/day for 4 weeks. No response rates were given, but the mean 17-item HRSD score decreased 6.2 points in week-8 non-responders and 10.1 points in partial responders. In a second study, Fava et al (Reference Fava, Rosenbaum and McGrath1994) randomised week-8 non-responders to fluoxetine 20 mg/day (n=41) to either fluoxetine 40–60 mg, desipramine addition or lithium addition for 4 weeks. No placebo increase was practised. Remission rates were 53%, 25% and 29%, respectively, but these differences were non-significant. Initial partial responders appeared to benefit most from fluoxetine dose increases (data non-significant). Drop-out rates for side-effects were 0%, 17% and 7%, respectively. In a third study, Fava et al (Reference Fava, Alpert and Nierenberg2002) repeated the three-arm randomised design from their 1994 study with a stratification for partial or non-response at week 8 (n=101). After 4 weeks, the high-dose fluoxetine group showed increased but non-significant remission rates (42.4%) compared with desipramine addition (29.4%) and lithium addition (23.5%). Again initial partial responders appeared to benefit more from fluoxetine dose increases compared with initial non-responders (differences non-significant). No specific data on drop-out because of side-effects were given.
Benkert et al (Reference Benkert, Szegedi and Wetzel1997) investigated dose escalation of paroxetine (20 mg/day) in out-patients who were depressed or had minor depression. Those who did not respond after 3 weeks of treatment (n=86) were randomised to receive 40 mg paroxetine for 3 additional weeks or placebo increase. Response rates were 75% in the placebo increased group and 74% in the 40 mg group. Licht & Qvitzau (Reference Licht and Qvitzau2002) investigated randomised dose escalation of sertraline (up to 200 mg/day) v. sertraline 100 mg/day (placebo increase) or mianserin addition in 295 outpatients non-responsive to sertraline 50 mg for 4 weeks and additionally increased to 100 mg for 2 more weeks. Response rates 5 weeks after randomisation were significantly lower in the dose-increase group (56%) than in the sertraline 100 mg group (70%) and the mianserin addition group (67%). Data on drop-out because of side-effects were not specified.
Strengths, flaws and other details of all selected studies are shown in Table 2. In summary, we mention several methodological problems we encountered: absence of placebo controls (Fava et al, Reference Fava, Cohen and Rosenbaum1992, Reference Fava, Rosenbaum and McGrath1994, Reference Fava, Alpert and Nierenberg2002), inclusion of minor depression (Reference Benkert, Szegedi and WetzelBenkert et al, 1997), insufficient data presentation (Reference Schweizer, Rickels and AmsterdamSchweizer et al, 1990; Reference Fava, Rosenbaum and McGrathFava et al, 1994), insufficient power (Schweizer et al, Reference Schweizer, Rickels and Amsterdam1990, Reference Schweizer, Rynn and Mandos2001; Fava et al, Reference Fava, Cohen and Rosenbaum1992, Reference Fava, Rosenbaum and McGrath1994, Reference Fava, Alpert and Nierenberg2002; Reference Benkert, Szegedi and WetzelBenkert et al, 1997), uncertainty about masking (Reference Dornseif, Dunlop and PotvinDornseif et al, 1989; Reference Schweizer, Rynn and MandosSchweizer et al, 2001), earlier dose escalation before the randomisation (Reference Licht and QvitzauLicht & Qvitzau, 2002), inadequate pooling of heterogeneous data and problems with conversion to dose equivalents (Reference Bollini, Pampallona and TibaldiBollini et al, 1999; Reference Baker, Tweedie and DuvalBaker et al, 2003). None of the studies provided information about the method of dose escalation or described the early drop-out rates because of dose escalation.
Evidence for dose escalation?
From four of the eight dose-escalation studies it appeared that dose increments before 4 weeks were not effective (level of evidence: A2) (Reference Dornseif, Dunlop and PotvinDornseif et al, 1989; Schweizer et al, Reference Schweizer, Rickels and Amsterdam1990, Reference Schweizer, Rynn and Mandos2001; Reference Benkert, Szegedi and WetzelBenkert et al, 1997; Reference Bollini, Pampallona and TibaldiBollini et al, 1999; Reference Corruble and GuelfiCorruble & Guelfi, 2000; Reference Baker, Tweedie and DuvalBaker et al, 2003). However, in the meta-analysis of some of these studies by Baker et al, a potential dose–response relationship was found for dose escalation if participants who dropped out because of side-effects were excluded from the analysis (a so-called dose-tolerant sample) (Reference Baker, Tweedie and DuvalBaker et al, 2003). Baker & Woods (Reference Baker and Woods2003) proposed that differential drop-out because of side-effects in the dose-escalation group (compared with placebo increase) conferred a substantial (negative) bias to the potential dose–response relationship. They argued that by applying a last-observation-carried-forward approach (often used in the original studies), more participants dropping out early (because of side-effects) in the high-dose groups would unequally increase average severity scores and decrease response rates compared with the lower-dose (or placebo) groups. This methodological problem could be overcome by analysing only dose-tolerant participants (those not dropping out because of side-effects).
In the well-performed study with sertraline by Licht & Qvitzau (Reference Licht and Qvitzau2002) (not included in the three reviews), dose escalation after 6 weeks was found to be less effective than continuation of the standard dose, or augmentation with mianserin (level of evidence: A2). After 8 weeks of treatment, increased dosages of fluoxetine were more effective than augmentation with lithium or desipramine (Fava et al, Reference Fava, Rosenbaum and McGrath1994, Reference Fava, Alpert and Nierenberg2002), although in the latter study this was not significant (level of evidence: B). In these studies no placebo dose escalation was performed. Both studies showed a non-significant trend of increased efficacy of dose escalation compared with augmentation (lithium or desipramine), particularly for partial responders (level of evidence: B).
Across all studies, higher doses were related to increased drop-out rates, which were associated with more side-effects in some studies (level of evidence: A2) (Reference Bollini, Pampallona and TibaldiBollini et al, 1999). It appeared that the occurrence of side-effects did not increase equally when dosages were gradually escalated for initial non-responders, compared with fixed-dose trials. However, this could not be compared straightforwardly between the studies, and was not investigated specifically.
Additional concerns for clinicians
We identified no evidence to recommend how dose increase should be practised. Also, the maximum dosage to be achieved was not investigated well.
DISCUSSION
Our systematic review provided eight studies about dose escalation of SSRIs. Only one of these studies approached our rather stringent criteria (Reference Licht and QvitzauLicht & Qvitzau, 2002). We found no evidence of increased efficacy by dose escalation within the first 4 weeks. Dose escalation after 6 weeks appeared less effective than continuing the same dose. We found some, but limited, evidence for efficacy of dose escalation after 8 weeks, particularly in partial responders. This effect was seen within 4 weeks after dose escalation. Irrespective of efficacy, dose escalation unequivocally increased side-effects, but effects on dropout rates because of side-effects were less straightforward. Thus, in the absence of methodologically well-designed studies we can neither unequivocally state that dose escalation is useful nor discard it as useless.
These findings may challenge the current beliefs and recommendations about dose escalation as it is generally practised (Reference Byrne and RothschildByrne & Rothschild, 1997; Reference Shergill and KatonaShergill & Katona, 1997; Reference Fredman, Fava and KienkeFredman et al, 2000; Reference Mischoulon, Nierenberg and KizilbashMischoulon et al, 2000). Contrary to this challenge, many patients who have only partially responded are too often treated with long-term obviously insufficient treatments (e.g. standard doses of SSRIs). For these patients, one could argue that it is better to try dose escalation than to continue inadequate treatment. Presumably, in the absence of clear guidance from trial data, clinicians do not have many alternatives for non-responders or partial responders, and clinicians all have their case histories of improvement after dose escalation. A more sophisticated question must therefore also be asked; i.e. which subgroup of patients will benefit from dose escalation?
So far, only the NICE guideline displayed some reserve in the general recommendation about dose escalation (National Institute for Clinical Excellence, 2004). The British Medicines and Healthcare products Regulatory Agency's Committee on Safety of Medicines examined the available evidence for dose escalation as provided by pharmaceutical companies, and recommended the lowest efficacious dose (Reference Weller, Ashby and BrookWeller et al, 2004). From this report it was unclear which studies were taken as evidence. Three previous reviews concerning higher doses of anti-depressants were published (Reference Bollini, Pampallona and TibaldiBollini et al, 1999; Reference Corruble and GuelfiCorruble & Guelfi, 2000; Reference Baker, Tweedie and DuvalBaker et al, 2003), the methodological shortcomings of which have already been mentioned. The findings in these reviews previously challenged the belief of a dose–response relationship, but Baker et al proposed a potential dose–response relationship, according to their dose-tolerance analysis. All reports summarised studies performed until 1997; thereafter, the study by Licht & Qvitzau (Reference Licht and Qvitzau2002) further challenged the efficacy of dose escalation.
Limitations of the identified studies
Four major issues of concern in the eight identified studies should be mentioned. First, the methodological quality of these studies varied between poor and good according to our classification. We summarised these methodological problems in the Results section and Table 2.
Second, and more in general, all dose-escalation studies, except the studies of Fava and colleagues, which lacked a placebo control (Fava et al, Reference Fava, Cohen and Rosenbaum1992, Reference Fava, Rosenbaum and McGrath1994, Reference Fava, Alpert and Nierenberg2002), suffered a methodological problem in the timing of dose escalation (Reference Baker and WoodsBaker & Woods, 2003). Even the most robust study, by Licht & Qvitzau (Reference Licht and Qvitzau2002), hampered its own design by a non-randomised dose increase 2 weeks before randomisation. This problem might explain the high placebo response rates in some of the dose-escalation studies (up to 75%).
Third, in most studies no data were provided on the selective drop-out, nor the schedule of dose increments (Reference Baker and WoodsBaker & Woods, 2003). The possibility that patients randomised to true dose escalation might drop out more frequently and earlier after randomisation (with associated high severity scores), compared with those receiving placebo, might have biased the intention-to-treat analyses in which last observations are usually carried forward to study end-points. This happens in particular when dose escalation is performed rapidly. The analysis of a dose-tolerant sample in such studies would indeed provide additional information, but these data were not provided.
Fourth, in the selected trials, it was mostly response that was used as primary outcome, whereas currently remission of depression is the clinical aim of treatment (Reference Nierenberg and DeCeccoNierenberg & DeCecco, 2001). If dose escalation would be effective, the question remains whether dose escalation will also further improve initial responders that were non-remitters. So far only Schweizer et al addressed this topic, with equivocal effects of dose escalation (Reference Schweizer, Rynn and MandosSchweizer et al, 2001).
Possible explanations for a dose–response relationship
A possible explanation of the clinical observation that response might occur after dose escalation is initial lower levels of the drug in the bloodstream. This may be related to increased metabolism because of genetic polymorphisms of the CYP enzyme system (Reference Bertilsson, Aberg-Wistedt and GustafssonBertilsson et al, 1985; Reference Steimer, Muller and LeuchtSteimer et al, 2001; Reference Charlier, Broly and LhermitteCharlier et al, 2003; Reference BrosenBrosen, 2004). The incidence of increased metabolism by (multi-)duplicated genes of CYP 2D6 varies between 1–2% in White populations in Sweden, 3.6% in Germany and 7–10% in Spain and Sicily, and also varies between ethnic groups (e.g. 29% in Black Ethiopians) (Reference Bertilsson, Dahl and DalenBertilsson et al, 2002). A few studies showed equivocal evidence for the involvement of CYP polymorphisms (responsible for rapid metabolism) as an explanation of non-response to a standard dose of SSRIs (Bertilsson et al, Reference Bertilsson, Dahl and Tybring1997, Reference Bertilsson, Dahl and Dalen2002; Reference Steimer, Muller and LeuchtSteimer et al, 2001; Reference BrosenBrosen, 2004; Reference Kawanishi, Lundgren and AgrenKawanishi et al, 2004). However, a clear relationship between blood levels of SSRIs and response was never found (Reference Beasley, Bosomworth and WernickeBeasley et al, 1990; Reference Norman, Gupta and BurrowsNorman et al, 1993; Reference BaumannBaumann, 1996; Reference Amsterdam, Fawcett and QuitkinAmsterdam et al, 1997; Reference Bourdeaux, Pannetier and YounosBourdeaux et al, 1998; Reference DeVaneDeVane, 1998). Perhaps genetic variability of the central target of these drugs, the serotonin reuptake transporter, may be responsible more directly for the effects of SSRIs (Reference Hahn and BlakelyHahn & Blakely, 2002; Reference Smits, Smits and SchoutenSmits et al, 2004).
From in-vitro and ex-vivo studies it appears that, at higher doses, selective anti-depressants such as SSRIs may become dual-action agents that, like noradrenaline, also affect other monoamine systems (Reference Owens, Morgan and PlottOwens et al, 1997; Reference Gorman and SullivanGorman & Sullivan, 2000; Reference Gilmor, Owens and NemeroffGilmor et al, 2002). From the current data on dose escalation in SSRIs, this theoretical hypothesis can neither be falsified nor proven. In addition, we are unaware of an acceptable method to test whether specific sites of action are responsible for the observed treatment effects.
Limitations of the review
No meta-analysis was performed because the differences in timing of dose escalation between the identified studies introduced substantial heterogeneity. An extension of the meta-regression approach as performed by Baker et al (Reference Baker, Tweedie and Duval2003) was considered inappropriate for addressing this problem, as the number of studies gave insufficient power; moreover, gender, age, outcome definition and type of SSRI ideally should be included in such a model.
The grading system for studies does not represent the appraised methodological dimensions of evidence. This improved the applicability of the results for busy clinicians, but reduced their strength.
Finally, patients studied in trials are generally selected populations, reducing external validity for clinical practice. All identified studies excluded psychotic depression, bipolar depression, depression in children or adolescents and depressive disorder with severe psychiatric and somatic comorbidity.
Future dose-escalation studies
For future dose-escalation trials, methodological issues should be considered. First, for optimal contrast in the study, an appropriate group of non-responders should be selected by postponing randomisation and refraining from (additional) interventions before dose escalation is applied. The minimum period that can be reconciled with recommendations in current guidelines and that is acceptable for clinical practice is 6 weeks. Second, studies should have enough power to detect significant differences. This implies a large sample to start with, as approximately 50% of patients will show a response in the first 6 weeks. Third, the method of dose escalation should be described and applied in such a way that few patients drop out. Fourth, adequate results should be presented: response and remission rates in intention-to-treat analyses and for the group that could be described as dose tolerant. Fifth, efficacy should be tested in predefined subgroups (e.g. partial responders at week 6). Sixth, genetic sampling (e.g. CYP and SERT polymorphisms) and plasma SSRI-level sampling would be interesting in the further examination of potential explanations for the clinically observed efficacy of dose escalation, and to identify potential prognostic variables.
Acknowledgements
This systematic review was realised by a grant for the development of a local evidence-based clinical practice guideline (No. SFA.07.012) from the Academic Medical Centre, Amsterdam, The Netherlands. This guideline considering strategies for non-response to a standard dose of a first SSRI is available from H.R. on request.




eLetters
No eLetters have been published for this article.