


Why electrochemical energy storage matters more than ever before
The recognition that energy can be stored at charged interfaces dates to the ancients: from borrowing the Greek word for amber (ηλεκτρον) to name the “electric ion,” electron; to the apparent electrochemical cell used over two millennia ago (the “Baghdad battery,” Figure 1a), which comprised an iron rod inserted into an electrolyte within a cylindrical copper vessel (all packaged in a clay pot) that functioned under load as a primary battery (discharge-only) as the Fe corroded; to the metal–air production of verdigris on copper by interspersing copper sheets with linens soaked in wine dregs. Batteries as a technology came of age during the scientific, technological, and intellectual fervor that characterized the Industrial Revolution. From Benjamin Franklin’s borrowing of the military term “battery” to designate Leyden jars (Figure 1b) connected in parallel to jolt 18th century experiments in electricity to life and amuse the curious in the salons of Europe―to the electric current generated by Volta’s pile of series-coupled Galvanic cells―to the chemicals (zinc, silver, lead) in use since the early 19th century to provide portable battery power. What can possibly be new under the sun from such a venerable―and practical―system?

Figure 1. (a) The terracotta-packaged object from over two millennia ago known as the Baghdad (or Parthian) battery; first proposed in 1940 as a power source to electroplate gold onto silver. Adapted from http://news.bbc.co.uk/2/hi/science/nature/2804257.stm. Note that devising low volume, hermetic, and lightweight packaging of the battery still bedevils the modern embodiment. (b) Photograph of a Leyden jar in which contact to a high surface-area electrified interface (gold foil) is made at the handle (electrode 1) versus the lead-composite toroidal electrode (electrode 2) painted on the exterior of the jar.
Not enough is the common lament. The Baghdad battery captures the necessary component parts of any battery or electrochemical capacitor: two physically separated electrodes of sufficient electronic conductivity with a medium for ion transport between them (Figure 1a). With technologists and consumers accustomed to the rate of improvement in computer operations known as Moore’s Law―paying half the cost every two years to double the number of operations per second―why should the performance of batteries trundle along with a mere 10% increase in energy density per year,Reference Hockenberry3, Reference Owens and Osaka4 taking a decade to double performance? The answer―physics versus electrochemistry―is both simple and complex. The metric of computation depends on the mobility of the electronic charge carrier, so performance improvements arise by optimizing this one functionality, typically by shortening the distance an electron or hole travels.
To store and release (charge/discharge) energy from a battery, three primary mobilities are involved: electronic transport in the solid state, ionic transport in the liquid and solid state, and molecular (mass) transport. If yearly performance improvement tracks at 1/2n, where n is the number of transport functions, integrated circuit computation has an n of 1 and doubles performance in two years, while n equals at least 3 for batteries for only a 12.5% improvement per year. Even in electrochemical double-layer capacitors in which an excess (or deficit) of electron charge on the electrode is balanced by a plane of counter-signed mobile ions (Figure 2), the frequency response of the two-electrode device and thus the power delivered is still dictated by electron mobility, ionic conductivity, and mass transport.
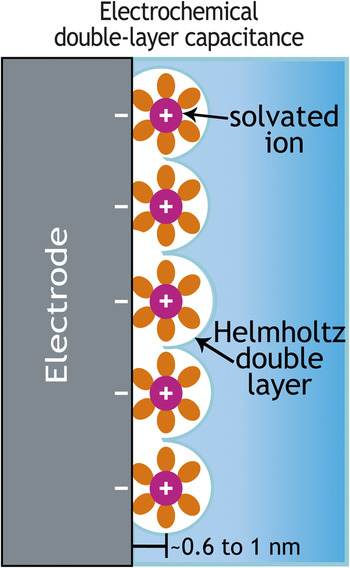
Figure 2. Schematic showing the spatially separated planes of charge that arise as oppositely signed mobile ions in the electrolyte balance the excess or deficit electronic charge at an electrified interface—thereby mimicking the spatially separated planes of positive and negative charge characteristic of an electrostatic capacitor.
As noted by Kurzweil and others, exponential growth of computational power (operations per second) pre-dates Moore’s Law and is a general law for electrical computation. The march of computational progress has been historically sustained because when the current state-of-the-art technology does not support an exponential rate of growth, a new set of hardware arises to do the computational work (e.g., discrete transistors replaced vacuum tube-based electronics and were, in turn, replaced by integrated circuits), as seen in Figure 3.Reference Kurzweil5

Figure 3. The exponential growth of computing over the 20th century: Calculations per second as a function of year from 1900–1998. Adapted from Reference Reference Kurzweil5. IC, integrated circuit; EM, electromagnetic.
Even though batteries in use today still employ materials and design concepts Volta and LeClanchéReference Reddy and Linden6 might recognize from 200 years ago, electrochemical energy storage has also experienced transitions to new performance curves. The battery chemistry powering one’s laptop has morphed in the past 20 years from nickel–cadmium (Ni–Cd) to nickel–metal hydride (NiMH) to lithium-ion (typically a graphitic carbon negative electrode versus a lithiated cobalt oxide positive electrode), fortunately becoming less environmentally problematic―and less heavy―in the process. Lithium-ion insertion materials, proposed by Whittingham in the mid-1970s as the active agent in the positive electrode,Reference Whittingham7 added the first new strategy in decades (if not centuries) to the portfolio of battery-derived portable power.
Electrochemical energy storage of the 21st century is similarly poised for a transition from the old to the new. Your father’s insertion battery is schematically shown in Figure 4. Whether a nonaqueous Li-ion or aqueous alkaline cell, the electrode is a composite structure with carbon powder, which provides an ad hoc electron wire into, throughout, and out of the electrode, bound in close contact with the poorly conductive powdered form of the active material that stores the potential energy of the inserted electron/cation charge (typically micrometer-sized LiCoO2 in Li-ion and micrometer-sized MnO2 in alkaline batteries). The composite electrode structure is simple in design and construction and economical to manufacture, as implied in Figure 4, yet in no way does it efficiently align and direct the transport functionality required for optimal energy storage: electron mobility, ion mobility, and molecular transport.
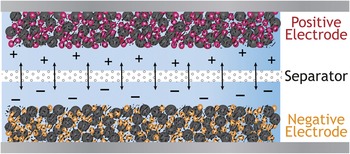
Figure 4. Schematic of your father’s (and your laptop’s) battery: Powder composites where the active material is blended with carbon powder to improve electron mobility through the composite structure.
Bringing a nanoscale perspective to the reaction interphases at which electrochemical energy is stored reveals another way to translate batteries and electrochemical capacitors onto a new performance curve. But the challenge is to move beyond mere creation and characterization of a functional nanoscale object or feature,Reference Long, Dunn, Rolison and White8–Reference Zhao, Mendoza Sánchez, Dobson and Grant10 because integrating transport functions of countervailing characteristics is essential in rate-critical applications such as energy storage.Reference Rolison, Long, Lytle, Fischer, Rhodes, McEvoy, Bourg and Lubers11 Materials design becomes complex when the structure of the material must accommodate order for high electron mobility, while ensuring ions and molecules have enough space to move to ensure high mobility. The research under way to transform your father’s battery into an advanced energy storage device that will play an integral role in the 21st century energy portfolio offers a blend of materials science, insight into nanoscale materials and phenomena, and re-wiring the transport paths necessary for power to hum.
Restructuring electrochemical energy storage: On the nanoscale and in three dimensions
Just as the reimagining of form and function on the nanoscale has propelled materials science, chemistry, and physics toward vital discoveries of technological relevance, electrochemical energy storage is already benefiting from a similar exercise. The nano-nexus for electrochemical energy storage science and engineering ranges from (1) retrieving materials from the historical discard pile, written off as materials of insufficient electronic conductivity or capacity, for a second look when expressed as nanomaterials; (2) stabilizing the physicochemical form of materials that yield multiple electrons per formula unit during conversion redox chemistry, thereby making them practical, active materials when nanoscopic; (3) recognizing that the solid product that accumulates in the air cathode of a lithium–air cell during discharge could be reversibly converted from lithium oxides to molecular oxygen in the presence of nanoscopic forms of catalytic manganese oxide, thereby sparking the battery equivalent of a gold rush, as research teams around the world hasten to study this system in order to create a rechargeable battery with the possibility of 10× the capacity of current lithium-ion batteries; (4) using the pseudocapacitance (redox-processes limited to the surface or near surface) of well-wired nanomaterials to move beyond the ca. 100 F g–1 of activated carbon-based electrochemical capacitors without relying on a nonaqueous electrolyte or a scarce platinum-group metal in hydrous ruthenium oxide to do the work of storing and releasing energy; to (5) integrating the active phases and their functions in three dimensions―part of the worldwide effort under way to take not just electrode structures but batteries into fully three-dimensionally integrated devices.
The clear advantages of nanosizing
The ability to scale electrode materials on the nanoscale can confer enormous advantages in rate enhancement (power) and storage capacity (energy density), with the caveat that the marked increases in surface area can also accelerate deleterious surface reactivity with the electrolyte. The adroit designer must particularly keep in mind that to ensure real benefits, a judicious choice of electrode material and electrolyte system is essential. Performance improvements can be significant, however. Inherent transport kinetics are typically not affected by nanosizing the active material, although even here, possibilities exist, owing to the potential of structural differences in nanomaterials. But nanometric reaction phases necessarily lead to both shorter transport distances and higher surface-to-volume ratios that improve redox accessibility and time for ion and electron transport versus the bulk material, owing to the familiar expression:
where τ is the transport time, l is the diffusion length, and D is the diffusion coefficient.
Hence higher utilization is attainable, and higher power rates are achievable. This downsizing in matter and upsizing in power has brought a range of negative electrode materials such as forms of titania (TiO2(B))Reference Armstrong, Armstrong, Canales, García and Bruce12 and Si (see Figure 5)Reference Hu, Wu, Hong, Cui, McDonough, Bohy and Cui13 closer to practical promise through the creation of nanowire 1D morphologies. Lithium titanate, Li4Ti5O12, is another material that benefits from control of the particle dimensions to the nanoregime,Reference Kim and Cho14 especially as the Li-ion diffusion rate is exceptionally high in this defect spinel lattice.Reference Takai, Kamata, Fujine, Yoneda, Kanda and Esaka15 Nanoscopic lithium titanate forms the negative electrode for numerous technologies, such as the Altair Nano cell. Similar principles, but on a multicomponent scale, resulted in the development of the Sn/C/Co composite that is now the negative electrode in Sony’s Nexelion cell that offers 30% more volumetric energy compared to conventional lithium-ion batteries. Recent work highlights these benefits for a new age of composite negative electrode materials comprising silicon and carbon (Si/C),Reference Wilson and Dahn16–Reference Cui, Yang, Hsu and Cui20 which rely on such design principles to achieve gravimetric capacities that approach 3× those of conventional carbons.
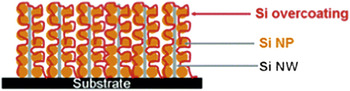
Figure 5. Schematic showing a network of silicon nanoparticle (NP)-decorated silicon nanowires (NWs)—a tailor-designed anode material for Li-ion batteries.Reference Hu, Wu, Hong, Cui, McDonough, Bohy and Cui13
Nanostructuring can also be cleverly utilized to create core-shell composite materials that can be tuned with respect to their outer and inner components to optimize performance. This strategy was used to make electrodes out of insulators; for example, a remarkable success story is illustrated by the cathode material LiFePO4.Reference Padhi, Nanjundaswamy and Goodenough21 These materials depend on coating thin conductive layers over the nanometric insulator in order to overcome their inherent low electronic conductivity, which otherwise impedes their practical exploitation. Formation of an intimate interface of the conductive phase with that of the active material is necessary. These composite nanostructures have been investigated intensively over the last decade, starting with the seminal report of Armand and colleagues on the carbon coating of olivine LiFePO4.Reference Ravet, Besner, Simoneau, Vallee, Armand and Magnan22 The most successful approach to increasing the specific power of the nanocomposite involves the design of nanodimensional (<100 nm) phosphate crystallites coated with a thin layer of carbon (an example is shown in Figure 6)―or ion conductive material―where the restricted dimensions also limit the number of crystalline defects that can inhibit transport in the 1D olivine tunnels.Reference Wang, Wang, Hosono, Wang and Zhou23, Reference Kang and Ceder24
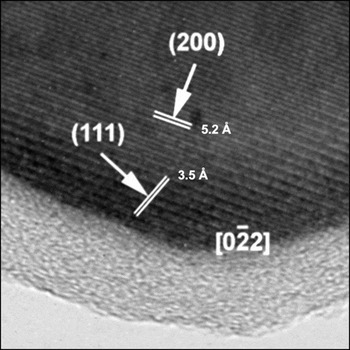
Figure 6. Transmission electron micrograph delineating the carbon coating on nanometric LiFePO4.
The interphase that forms between the nanoscale active material in the electrode and the electrolyte also forms a complex nanostructure that can be tailored to optimize transport properties. Another advantage presented by the use of such nanocrystallite composites is the increased electrode/electrolyte surface area available for Li+ access. This surface-to-volume amplification can enhance ion-transfer rates across the surface layer by many orders of magnitude and enable ultrahigh rate behavior. Hundreds of papers have been devoted to the enhancement of capacity and power through such deliberate engineering, not only for LiFePO4, but also for other members of the olivine family and other phosphate and fluorophosphate structures.Reference Song, Lee, Kim, Nazar and Cho25 Because an accompanying drawback of nanostructuring is to enhance detrimental surface reactivity with the electrolyte, this design strategy was deemed best suited to materials of intermediate equilibrium potentials (i.e., neither strongly oxidizing materials for the cathode nor strongly reducing materials at the anode). Lower cell voltages are a consequence―as are safer cells that reduce the risk of battery venting, fire, and explosion.
A different twist on core–shell composites has been used recently to design lithiated metal-oxide composites that express high cathode potentials. The article by Chen et al. in this issue discusses a new exciting family of cathode materials that employs a graded metal-oxide nanostructure that places the less surface-reactive oxide at the outer surface and the higher capacity oxide in the particle interior. These functional gradients enable significant improvements in capacity, energy density, and especially cycling stability.
Arguably, the battery systems that can most benefit from nanochemistry, however, are those that advance “beyond Li-ion” and beyond the intercalation chemistry that serves as the basis for the batteries described previously. Because the redox chemistry of the positive electrodes in such materials is coupled with substantial morphological/structural dynamics, nanostructured materials have a particularly crucial role to play. The functioning of the electrodes completely relies on the intimate contact of reactants and products that nanostructuring can provide, as well as the improved mechanical flexibility of nanomaterials relative to their bulk form. Such systems are important because Li-ion batteries are approaching the energy-density limits that intercalation materials can potentially provide, about 300 mA h g–1. At present, battery technology poses severe limitations on the driving range of electric or plug-in hybrid vehicles. New systems are being sought for next-generation batteries to provide much higher energy densities and reduce cost factors. The most favorable involve the electrochemical reactions that provide the basis for Li–S and Li–air batteries. The Li–S battery operates by reversible reaction of elemental sulfur at the positive electrode with lithium to form Li2S; a nanostructured carbon host serves to not only form intimate electronic contact with the insulating sulfur mass, but also to constrain the reaction products by providing a tortuous path that limits the loss of polysulfide intermediates by diffusion .Reference Ji, Lee and Nazar26, Reference Liang, Dudney and Howe27 The Li–air cathode (shown in Figure 7) is similar to that of a fuel cell. Oxygen from the atmosphere is supplied to the electrode and reversibly reduced to form Li2O2in situ. The reaction at the negative electrode is based on oxidation of Li (to Li+ + e–) instead of the oxidation of a fuel such as H2 or hydrocarbons. The energy density is extremely high compared to that of other rechargeable systems―up to 2–3 kWhkg–1 with respect to the mass of the discharged electrode.
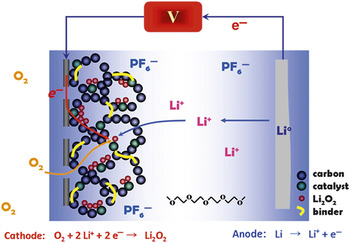
Figure 7. Schematic of a Li–air battery.
But in practice, the Li–air system presents tremendous challenges. The metal peroxide generated upon discharge is stored within the voids of a porous carbon that acts as a membrane, catalyst support, and co-catalyst; both pore architecture and volume are of paramount concern, since these factors limit the electron storage capacity. On charge, Li2O2 is converted back to Li and O2, where intimate contact with the carbon support―and a catalyst such as metal oxide or platinum-group metal nanoparticle―is vital. The article by Bruce et al. in this issue summarizes the recent developments in Li–air batteries and Li–S batteries, which share many operational and design concepts in common as well as important future challenges.
Use of deliberately defective nanoscale materials to enhance capacity—Something from nothing
The presence of defects and vacancies strongly influences the properties of transition metal oxides,Reference Goodenough28, Reference Cox29 including electrochemical capacity, electronic conductivity, electrode potential, and ionic transport. Ruetschi previously demonstrated the role of cation vacancies in proton-insertion reactions at electrolytic MnO2 (EMD) electrodes and explained the thermodynamics behind the curious fact that an absence of metal centers (the Mn4+ cations) (Figure 8) increased the cell voltage of the alkaline cell (Zn||KOH||EMD).Reference Ruetschi30–Reference Ruetschi and Giovanoli32

Figure 8. A depiction of Mn4+ cation vacancies that arise in the ramsdellite–pyrolusite intergrowth defect structure for γ-MnO2 (electrolytic MnO2, EMD); these vacancies are proton-stabilized.
Atypical insertion capacities reappeared with the expression of V2O5 in aerogel form.Reference Chaput, Dunn, Fuqua and Salloux33, Reference Le, Passerini, Guo, Ressler, Owens and Smyrl34 The V2O5 aerogels possess the characteristic ribbon morphology of V2O5 gels but are a low-density expression of the material fully continuous with the mesoporous network of the ultraporous aerogel nanoarchitecture. Where the aerogel form truly distinguishes itself from the character of polycrystalline or xerogel forms of V2O5 is in the number of Li/V2O5 inserted, with ≥ 2 Li/V reported by both groups.Reference Chaput, Dunn, Fuqua and Salloux33, Reference Le, Passerini, Guo, Ressler, Owens and Smyrl34 Note that when the stoichiometry reaches 4, 5, or 6 Li per V2O5, it is not clear that insertion (or intercalation) is the correct description of the Li–V2O5 storage process.Reference Rolison and Dunn35 The ability of V2O5 aerogels to insert Mg2+, Al3+, and Zn2+ also signaled that this form of the oxide offered more elbow room to host cations that do not insert into the polycrystalline or xerogel forms of the oxide.Reference Le, Passerini, Coustier, Guo, Soderstrom, Owens and Smyrl36
To help resolve the nature of the increased Li-ion capacity obtained with V2O5 in aerogel form, Swider-Lyons et al. turned to classic solid-state-ionics protocol to induce various point defects in micrometer-sized V2O5 polycrystalline powders.Reference Swider-Lyons, Love and Rolison37 Various temperature/atmosphere treatments were used to control the nature of vacancies within the initial orthorhombic crystalline habit, which significantly affected the Li-ion capacities of the treated powders as measured in a standard powder composite electrode structure, Figure 9. Oxygen (anion) vacancies and Schottky defects (anion and cation vacancies) lowered the lithium-ion capacity relative to composite electrodes prepared with the as-received polycrystalline material. The only treatment that increased Li-ion capacity―by 23%―was the creation of proton-stabilized cation vacancies, as formed by roasting the polycrystalline powder in an O2/H2O atmosphere.

Figure 9. Charge-discharge profiles in 1 M LiClO4/propylene carbonate of a powder composite electrode structure containing 0.5 mg of 1-μm polycrystalline V2O5 treated at 460°C in different atmospheres, as shown. (a) First complete charge cycle (10 μA). (b) First complete discharge cycle (10 μA). The only treatment that increases Li-ion capacity in the bulk oxide above that of the as-received material is to create proton-stabilized cation vacancies by heating in an O2/H2O atmosphere. Reprinted with permission from Reference Reference Swider-Lyons, Love and Rolison37. ©2002, Elsevier.
Hahn et al. recently reported that by creating proton-stabilized cation vacancies in iron oxide, a cost-effective and naturally abundant compound, they could tailor a rejected cathode material (γ-Fe2O3, maghemite) to one that achieved technologically relevant Li-ion storage capacities of >100 mAhg–1 at potentials above 2.0 V versus Li/Li+, making it a practical cathode material for Li-ion batteries.Reference Hahn, Long, Pettigrew, Osofsky and Rolison38 Rather than roasting the native ferrite in an O2/H2O atmosphere, substituting Mo6+ for octahedral-sited Fe3+ generated a large population of Fe3+ vacancies (∼0.9 transition metal cation vacancies per formula unit). In analogy with Ruetschi’s results, these Mo6+-induced, proton-stabilized cation vacancies increased the Li-ion storage relative to the native ferrite (>100 mA h g–1 versus ∼16 mA h g–1 for the unsubstituted ferrite control) and increased the electrochemical potential of the oxide to move the capacity into a cathode-relevant range of 4.1 and 2.0 V.
Dual function in one nanostructured electrode: Blended battery and electrochemical capacitor performance
The Ragone plot, which contrasts the specific power and specific energy of power source devices, spans the application space from the instant gratification of dielectric capacitors (power *now*!) to the reactant-fed endurance of fuel cells (Figure 10). Electrochemical capacitors and batteries bridge the energy/power/time spectrum between them. While a Ragone plot orients one when selecting the best power source for the job (or mission) at hand, it also imposes a restricted view―bordering on dogma―of the energy-storage possibilities. One simply did not look to electrochemical capacitors for energy density or to batteries for power density. But preparing charge-storage materials in nanoscale form―and especially wiring insertion materials in 3D―is blurring the distinction between battery-only character and pulse-power-only character. This blending of function makes innately hybrid devices feasible and allows electrochemical storage devices to explore the upper-right quadrant of the Ragone plot, as discussed for asymmetric capacitors by Long et al. and for 3D strategies by Arthur et al. in this issue.
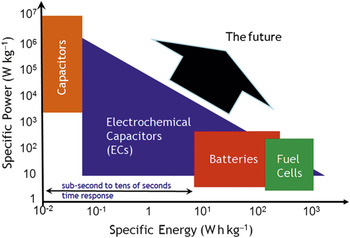
Figure 10. Instant versus delayed gratification: The comparison of specific power and specific energy of electrical energy-storage devices known as a Ragone plot.
Work by Martin and coworkers 15 years ago demonstrated that templated 1D forests of nanometric charge-insertion materials, such as Li2MnO4 and TiS2, behaved comparably to microstructured thin films of the material at slow discharge rates (C/20, where a 1C rate equals full discharge of the battery capacity in one hour) but could provide more capacity at high discharge rates.Reference Nishizawa, Mukui, Kuwabata, Martin and Yoneyama39, Reference Che, Jirage, Fisher and Martin40 For template-synthesized V2O5, the nanostructured tendrils provided fourfold more capacity at rates >500 CReference Patrissi and Martin41 and could still deliver capacity at high discharge rates at temperatures below 0°C, where batteries can have limited capability.Reference Sides and Martin42 Electrochemical studies of electrically conductive oxide aerogels, where the feature size is on the order of 10 nm, helped demonstrate that insertion oxides on the nanoscale possess both battery-like and capacitor-like behavior.Reference Rolison and Dunn35 Smyrl and coworkers used coulometric titration to demonstrate that V2O5 aerogels exhibited two voltage–time (V–t) domains with capacitor-like behavior at short times (V ∝ t) for the portions of the insertion material in closest contact to the current collector, which then transitioned to diffusion-limited character (V ∝ √t) at longer times.Reference Coustier, Lee, Passerini and Smyrl43 It was then shown that abandoning the classic powdered composite electrode structure to formulate a monoparticulate layer of V2O5 aerogel in direct contact with its current collector creates an electrode structure with explicit coupling of capacitive (EC) and faradaic (battery) behavior.Reference Dong, Dunn and Rolison44
Long et al. have converted these observations into practical form by designing electrode structures via electroless deposition of birnessite-like manganese oxide (MnOx) as ∼10-nm thick layers on the high-surface-area walls of 3D ultraporous carbon nanofoam papers.Reference Long, Fischer and Rolison45 These papers are device-ready electrodes that no longer require conductive additives and polymer binders to convert the nanomaterials into a usable macroscale object.Reference Long and Rolison46 In alkaline electrolyte,Reference Long, Sassin, Fischer, Rolison, Mansour, Johnson, Stallworth and Greenbaum47 the oxide–carbon nanofoam composite provides both pulse power (on the order of a few Farads per cm–2) and reversible, faradaic battery chemistry at insertion potentials (Figure 11). The intimate wiring of the insertion oxide to its massively parallel 3D current collector (the carbon nanofoam) also yielded reversible cycling in an aqueous electrolyte of 0.7 electrons per Mn center rather than ≤0.5 electrons per Mn center. Structural analysis by extended x-ray absorption fine-structure demonstrated that no structural conversion occurred during the capacitive portion of the voltammetry, but in the battery portion of the curve, the structure of the layered birnessite converted to MnOOH upon discharge, with structure recovery upon recharge.Reference Long, Sassin, Fischer, Rolison, Mansour, Johnson, Stallworth and Greenbaum47 Melding nanoscopic insertion oxide plus intimate wiring to the current collector, along with the 3D amplification necessary to deliver more than miniscule amounts of energy or power, creates new terrain: an aqueous rechargeable manganese oxide electrode that can store/release more than 0.5 e–/Mn.
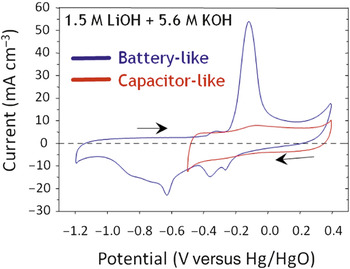
Figure 11. Ability of MnOx-painted carbon nanofoam paper to perform with both electrochemical capacitor character and battery response. In battery mode, repeated structural and phase interconversion of layered birnessite-like MnOx and MnOOH is obtained on deep charge–discharge with 0.7 e–/Mn reversibly cycled over 25 charge–discharge cycles in the aqueous electrolyte. Adapted from Reference Reference Long, Sassin, Fischer, Rolison, Mansour, Johnson, Stallworth and Greenbaum47. ©2009, American Chemical Society.
Summary
A unifying theme in this article is the principle of REsearch, where old ideas, brought back to the table but in a new light, enable step changes in the science and technology of energy storage. The last decade has witnessed many such developments involving a plethora of nanostructured materials prepared by deliberate design, which are advantageous for enhancing electronic, ionic, and molecular transport in electrode materials and structures. What we are starting to realize is that in contrast to attempting to tailor a single material, nanostructured materials, composites, and architectures can provide an optimum way of crafting desired characteristics for a target application. The approach allows one to physicochemically adjust the material to suit the application need or to compensate for the limitations inherent within one material alone. Approaches that have been used to blend the structures at the nanometer level have been remarkably successful, and we eagerly anticipate the developments of the next decade that may be inspired by the articles within this issue.
Acknowledgments
The authors acknowledge the support of the U.S. Office of Naval Research (D.R.R.) and the Natural Science and Engineering Research Council of Canada (L.F.N.) for their respective programs in nanomaterials and electrochemical energy storage. The authors also thank Megan B. Sassin of the U.S. Naval Research Laboratory for preparing Figures 2 and 4.











