



Introduction
The Ce-dominant rare earth element (REE) phosphate mineral monazite-(Ce) is the most commonly occurring REE-bearing mineral and most abundant species of the monoclinic monazite group. It occurs typically as a small accessory phase in granitic and metamorphic rocks, but reaches larger sizes and higher modal abundancies in some carbonatites, pegmatites, alkaline rocks and metamorphic veins (e.g. Bermanec et al., Reference Bermanec, Tibljaš, Gessner and Kniewald1988; Andreoli et al., Reference Andreoli, Smith, Watkeys, Moore, Ashwal and Hart1994; Lehmann et al., Reference Lehmann, Nakai, Höhndorf, Brinckmann, Dulski, Hein and Masuda1994; Wall and Mariano, Reference Wall, Mariano, Jones, Wall and Williams1996; Gonçalves et al., Reference Gonçalves, Lana, Scholz, Buick, Gerdes, Kamo, Corfu, Marinho, Chaves, Valeriano and Nalini2016; Broom-Fendley et al., Reference Broom-Fendley, Wall, Spiro and Ullmann2017; Montel et al., Reference Montel, Razafimahatratra, de Parseval, Poitrasson, Moine, Seydoux-Geuillaume, Pik, Arnaud and Gibert2018; Ntiharirizwa et al., Reference Ntiharirizwa, Boulvais, Poujol, Branquet, Morelli, Ntungwanayo and Midende2018; Slezak and Spandler, Reference Slezak and Spandler2019). Monazite-(Ce) is of considerable importance for both practical and research reasons. It is a major ore mineral for the light (L)REE (and Th) (Chakhmouradian and Wall, Reference Chakhmouradian and Wall2012; Wall, Reference Wall and Gunn2014; Cuney and Kyser, Reference Cuney, Kyser, Cuney and Kyser2015), forming high grade ‘hard rock’ deposits such as Steenkampskraal, South Africa (Andreoli et al., Reference Andreoli, Smith, Watkeys, Moore, Ashwal and Hart1994) and Kangankunde, Malawi (Wall and Mariano, Reference Wall, Mariano, Jones, Wall and Williams1996), as well as forming in secondary environments such as laterites (e.g. Mt Weld, Western Australia; Lottermoser, Reference Lottermoser1990) and heavy mineral sands, both of which are mined. It is also a potential host for long-term storage of radioactive waste (Ewing and Wang, Reference Ewing, Wang, Kohn, Rakovan and Hughes2002; Oelkers and Montel, Reference Oelkers and Montel2008), due to its ability to structurally incorporate a variety of actinide elements and resistance to metamictisation (Seydoux-Guillaume et al., Reference Seydoux-Guillaume, Deschanels, Baumier, Neumeier, Weber and Peuget2018).
Monazite-group minerals are used widely as petrochronometers (Engi, Reference Engi, Kohn, Engi and Lanari2017). Both Th and U are incorporated readily into the monazite structure, while common Pb concentrations are typically low, enabling Th–Pb and, when U contents are sufficiently high, U–Pb age determination using isotopic (e.g. Parrish, Reference Parrish1990) and non-isotopic techniques such as by electron-probe microanalysis (EPMA; Montel et al., Reference Montel, Foret, Veschambre, Nicollet and Provost1996; Williams et al., Reference Williams, Jercinovic and Hetherington2007). Moreover, diffusion of heavy elements is very slow (Cherniak et al., Reference Cherniak, Watson, Grove and Harrison2004; Gardés et al., Reference Gardés, Jaoul, Montel, Seydoux-Guillaume and Wirth2006, Reference Gardés, Montel, Seydoux-Guillaume and Wirth2007), meaning that inherited cores and individual zones in texturally complex monazite may preserve conditions from multiple, temporally distinct, growth events (Hetherington et al., Reference Hetherington, Backus, McFarlane, Fisher, Pearson, Moser, Corfu, Darling, Reddy and Tait2018). Combining geochronological data with other geochemical, isotopic and textural details means that the history of rocks formed over multiple episodes can be unravelled (Engi et al., Reference Engi, Lanari, Kohn, Kohn, Engi and Lanari2017). For example, monazite-(Ce) in equilibrium with xenotime-(Y) (Spear and Pyle, Reference Spear, Pyle, Kohn, Rakovan and Hughes2002) may be used as a thermometer, constraining P–T–t conditions. Combining δ18O (e.g. Ayers et al., Reference Ayers, Loflin, Miller, Barton and Coath2006) or 143Nd/144Nd (e.g. McFarlane and McCulloch, Reference McFarlane and McCulloch2007) with Th–Pb age determination can trace hydrothermal fluid infiltration and mixing between different melt or fluid sources. Textural features, such as oscillatory and sector zoning (e.g. Cressey et al., Reference Cressey, Wall and Cressey1999) may be attributed to magmatic crystallisation, resorption may indicate the presence of a melt with which monazite-(Ce) is not in equilibrium, while development of mineral inclusions, porosity, or mottled textures may be indicative of fluid-mediated dissolution–reprecipitation and therefore ingress of external fluids (Putnis, Reference Putnis2002, Reference Putnis, Oelkers and Schott2009; Hetherington et al., Reference Hetherington, Backus, McFarlane, Fisher, Pearson, Moser, Corfu, Darling, Reddy and Tait2018).
While rare, the substitution of sulfur into monazite-(Ce) has been described in several localities, predominantly in carbonatites (e.g. Kukharenko et al., Reference Kukharenko, Bulakh and Baklanova1961; Cressey et al., Reference Cressey, Wall and Cressey1999; Bulakh et al., Reference Bulakh, Nesterov, Zaitsev, Pilipiuk, Wall and Kirillov2000; Wall, Reference Wall and Vladykin2004; Enkhbayar et al., Reference Enkhbayar, Sea, Choi, Lee and Batmunkh2016; Prokopyev et al., Reference Prokopyev, Doroshkevich, Ponomarchuk and Sergeev2017; Nikolenko et al., Reference Nikolenko, Redina, Doroshkevich, Prokpyv, Rafozin and Vladykin2018), but also in kimberlites (Chakhmouradian and Mitchell, Reference Chakhmouradian and Mitchell1999), fluid-altered rhyolite (Ondrejka et al., Reference Ondrejka, Uher, Pršek and Ozdín2007) and in metamorphic rocks (Suzuki and Kato, Reference Suzuki and Kato2008; Pršek et al., Reference Pršek, Ondrejka, Bačík, Budzyń and Uher2010; Krenn et al., Reference Krenn, Putz, Finger and Paar2011; Ondrejka et al., Reference Ondrejka, Putiš, Uher, Schmiedt, Pukančík and Konečný2016; Laurent et al., Reference Laurent, Seydoux-Guilaume, Duchene, Bingen, Bosse and Datas2016). Small amounts of sulfur in monazite-(Ce) and monazite-(La) are also sometimes noted during EPMA radiometric age determination, owing to the close proximity of the SKα line to the PbMα lines (Jercinovic and Williams, Reference Jercinovic and Williams2005; Suzuki and Kato, Reference Suzuki and Kato2008), as well as the association of high S contents with increased common Pb (Krenn et al., Reference Krenn, Putz, Finger and Paar2011). Early work predominantly explored substitution mechanisms for incorporating sulfur in monazite-(Ce) (e.g. Chakhmouradian and Mitchell, Reference Chakhmouradian and Mitchell1999; Bulakh et al., Reference Bulakh, Nesterov, Zaitsev, Pilipiuk, Wall and Kirillov2000; Ondrejka et al., Reference Ondrejka, Uher, Pršek and Ozdín2007), but the presence of sulfur is being used increasingly to infer formation environments. Sulfur incorporation is assumed generally to reflect the introduction of S-bearing fluids in relatively acidic and high ![]() $f_{O_2}$ conditions (Pršek et al., Reference Pršek, Ondrejka, Bačík, Budzyń and Uher2010; Krenn et al., Reference Krenn, Putz, Finger and Paar2011; Ondrejka et al., Reference Ondrejka, Putiš, Uher, Schmiedt, Pukančík and Konečný2016; Laurent et al., Reference Laurent, Seydoux-Guilaume, Duchene, Bingen, Bosse and Datas2016), possibly at temperatures lower than 400°C based on monazite-(Ce)–xenotime-(Y) thermometry (Krenn et al., Reference Krenn, Putz, Finger and Paar2011). Recently, Laurent et al. (Reference Laurent, Seydoux-Guilaume, Duchene, Bingen, Bosse and Datas2016) showed that it is possible to precisely date S incorporation in monazite-(Ce) and therefore understand the timing of S mobilisation in metamorphic rocks. Their study highlighted the potential to date S-rich domains in monazite-(Ce) from other rock types, such as ore deposits, to further understand the timing of mineralisation. On the basis of co-crystallisation of baryte and the absence of sulfides (e.g. Ondrejka et al., Reference Ondrejka, Uher, Pršek and Ozdín2007, Reference Ondrejka, Putiš, Uher, Schmiedt, Pukančík and Konečný2016), it is generally assumed that monazite-(Ce) only incorporates S as the S6+ ion, and therefore the presence of S-bearing monazite-(Ce) reflects S mobility in oxidising conditions only. However, experimental and ab initio studies have shown that apatite, which has been assumed typically to accommodate sulfur as S6+ only (e.g. Pan and Fleet, Reference Pan, Fleet, Kohn, Rakovan and Hughes2002), is also capable of accommodating more reduced varieties of S such as S4+ and S2– (Konecke et al., Reference Konecke, Fiege, Simon, Parat and Stechern2017a; Kim et al., Reference Kim, Konecke, Fiege, Simon and Becker2017). There has been no direct investigation into the redox state of S in monazite-(Ce) to investigate its capability to accommodate S in different oxidation states. Moreover, the substitution mechanism incorporating S in monazite-(Ce) remains incompletely understood. These two less-constrained factors reduce our confidence for inferring the formation environment from the presence of S in LREE phosphate species of the monazite group.
$f_{O_2}$ conditions (Pršek et al., Reference Pršek, Ondrejka, Bačík, Budzyń and Uher2010; Krenn et al., Reference Krenn, Putz, Finger and Paar2011; Ondrejka et al., Reference Ondrejka, Putiš, Uher, Schmiedt, Pukančík and Konečný2016; Laurent et al., Reference Laurent, Seydoux-Guilaume, Duchene, Bingen, Bosse and Datas2016), possibly at temperatures lower than 400°C based on monazite-(Ce)–xenotime-(Y) thermometry (Krenn et al., Reference Krenn, Putz, Finger and Paar2011). Recently, Laurent et al. (Reference Laurent, Seydoux-Guilaume, Duchene, Bingen, Bosse and Datas2016) showed that it is possible to precisely date S incorporation in monazite-(Ce) and therefore understand the timing of S mobilisation in metamorphic rocks. Their study highlighted the potential to date S-rich domains in monazite-(Ce) from other rock types, such as ore deposits, to further understand the timing of mineralisation. On the basis of co-crystallisation of baryte and the absence of sulfides (e.g. Ondrejka et al., Reference Ondrejka, Uher, Pršek and Ozdín2007, Reference Ondrejka, Putiš, Uher, Schmiedt, Pukančík and Konečný2016), it is generally assumed that monazite-(Ce) only incorporates S as the S6+ ion, and therefore the presence of S-bearing monazite-(Ce) reflects S mobility in oxidising conditions only. However, experimental and ab initio studies have shown that apatite, which has been assumed typically to accommodate sulfur as S6+ only (e.g. Pan and Fleet, Reference Pan, Fleet, Kohn, Rakovan and Hughes2002), is also capable of accommodating more reduced varieties of S such as S4+ and S2– (Konecke et al., Reference Konecke, Fiege, Simon, Parat and Stechern2017a; Kim et al., Reference Kim, Konecke, Fiege, Simon and Becker2017). There has been no direct investigation into the redox state of S in monazite-(Ce) to investigate its capability to accommodate S in different oxidation states. Moreover, the substitution mechanism incorporating S in monazite-(Ce) remains incompletely understood. These two less-constrained factors reduce our confidence for inferring the formation environment from the presence of S in LREE phosphate species of the monazite group.
This study presents the first description of S-bearing monazite-(Ce) from the Eureka carbonatite, Namibia. Eureka exhibits the largest grains of S-bearing monazite-(Ce) yet documented (>0.5 mm) with an SO3 concentration within uncertainty of the highest published SO3 contents [11.23 wt.% SO3 ≡ 0.294 S6+ atoms per formula unit (apfu); cf. Pršek et al., Reference Pršek, Ondrejka, Bačík, Budzyń and Uher2010]. The large size of the S-bearing monazite-(Ce) has enabled analysis by X-ray photoelectron spectroscopy (XPS), and we use this technique to present the first direct data on the oxidation state of S in monazite-(Ce). We also expand on previous work investigating S substitution in monazite-(Ce), moving towards a complete, charge-balanced, understanding of the S substitution mechanism. Lastly, on the basis of the crystallisation environment, we show that sulfur can be incorporated into monazite-(Ce) during a protracted weathering process, opening up the possibility of directly dating palaeo-weathering horizons.
Geology of the Eureka carbonatite
The Eureka carbonatite is located at 22.044°S, 15.254°E on the property of Eureka Farm 99, near Usakos, Namibia (Fig. 1a). Quaternary gravels and calcrete obscure much of the exposure, but remnant mineral exploration trenches from the late 1980s, and modern trenching by E-Tech Metals, facilitates easy sampling. The carbonatites comprise a series of at least four steeply-dipping (>75°) monazite-(Ce)-bearing dolomite–carbonatite dykes and at least three monazite-(Ce)-poor dolomite carbonatites and sövites, all striking roughly parallel, (Fig. 1b; von Knorring and Clifford, Reference von Knorring and Clifford1960; Dunai, Reference Dunai1989). The width of the dykes is typically 1–2 m, but may locally reach up to 7 m. The dykes intrude parallel to the foliation of intensely folded Damaran schists and quartzites of the Etusis formation (Miller, Reference Miller and Miller1983, Reference Miller2008). An 87Sr/86Sr isotope study supports a mantle origin, with minor crustal input (Dunai et al., Reference Dunai, Stoessel and Ziegler1989).
Fig. 1. Location map (a) and geological sketch map (b) of the Eureka carbonatite dykes, with pit and sample locations. Host rocks are quartzite and schists of the Etusis formation. Geological map grid is UTM 33S, WGS 1984 datum, redrawn and georeferenced from Dunai (Reference Dunai1989).
A distinct feature of the dykes at Eureka is the abundance and size of monazite-(Ce) in the carbonatite. Grains reach up to 30 cm in size and have been divided by Dunai (Reference Dunai1989) by morphology, where (more abundant) larger grains are typically euhedral to subhedral while smaller grains are lenticular and display resorption features. Monazite-(Ce) from Eureka has been dated, by U–Pb, as 500 ± 20 Ma (Burger et al., Reference Burger, von Knorring and Clifford1965) and 548 ± 4 Ma (Ragettli et al., Reference Ragettli, Hebeda, Signer and Wieler1994; Gonçalves et al., Reference Gonçalves, Lana, Scholz, Buick, Gerdes, Kamo, Corfu, Rubatto, Wiedenbeck, Nalini and Oliveira2018).
Extensive trenching indicates that the area is weathered to at least 2 m and carbonatite dykes are locally replaced by an assemblage of secondary carbonate minerals, gypsum, serpentine(?), iron oxide/hydroxides, clay and minor fluorite. Locally, the carbonatite is intensely silicified; silicification is most prominent in the north-western area of the intrusion (Zone 2 of Dunai, Reference Dunai1989), with recent drilling indicating that silicification extends to a maximum probable depth of ~5 m. In these rocks, the carbonate matrix has been completely replaced by iron-bearing, rust-brown chalcedony (Fig. 2). Remnant monazite-(Ce) grains in the silicified rocks are typically, although not always, rimmed by a thin (up to 5 mm) alteration halo of S-bearing monazite-(Ce), which is the subject of this paper (Fig. 2). The local area is commonly capped by a layer of calcrete ~30 cm thick.
Fig. 2. Example of monazite-(Ce)-bearing (Mnz) silicified carbonatite from Eureka. Note the ~500 μm S-bearing monazite-(Ce) rims (S-Mnz) around the monazite-(Ce) grains.
Samples and methods
Representative silicified carbonatite samples were collected from Pit 2, in the north-western zone of the deposit (Fig. 1b), where silicification is most intense. Petrographic observations were undertaken using conventional polarising microscopy and back-scattered electron (BSE) imaging. BSE images and maps from energy-dispersive spectroscopy (EDS) were obtained using a FEI Quanta 650 FEG SEM hosted at the Environment and Sustainability Institute, University of Exeter.
Electron probe microanalyses were carried out at Camborne School of Mines on a JEOL JXA-8200 Superprobe, using a defocussed 10 μm, 15 kV, 50 nA beam. Peak counting times were 20 s for S, Fe, P, Ca and Sr; 30 s for F, Na, Ba and Ce; 40 s for Si, Nd, La, Pr and Sm; 60 s for Th; 100 s for Y; and 120 s for Gd. Background count times were half those of the peak value. X-ray counts were converted to wt.% oxide using the in-built JEOL CITZAF correction program. Empirical interference corrections were performed for the REE following Williams (Reference Williams, Jones, Wall and Williams1996). Correction factors were calculated by measuring the interferences observed on individual REE reference standards. Commercial (Astimex) natural minerals standards were used for all elements except for Th and the REE, where Astimex Th metal and REE–Si–Al–Ca–O glasses from the University of Edinburgh, were used. Representative detection limits are included in Supplementary Table S1.
Laser ablation inductively-coupled plasma mass spectrometry (LA-ICPMS) was carried out at the University of Leeds, using a Lambda Physik 193 nm ArF Excimer laser coupled to an Agilent 7500c ICPMS. This was run in reaction mode with 2.5 ml/min H2 in the reaction cell. The laser utilised a 10 Hz, 25 μm spot for ~30 s of ablation time. Typical fluence was 10 J cm–2. Isotopes analysed were: 23Na, 28Si, 31P, 32S, 40Ca, 47Ti, 51V, 56Fe, 75As, 88Sr, 89Y, 90Zr, 93Nb, 65Mo, 137Ba, 139La, 140Ce, 141Pr, 147Sm, 153Eu, 157Gd, 159Tb, 163Dy, 165Ho, 166Er, 169Tm, 172Yb, 175Lu, 178Hf, 208Pb, 232Th and 238U. Data were processed using Iolite software (version 2.5; Paton et al., Reference Paton, Hellstrom, Paul, Woodhead and Hergt2011). Neodymium contents from the EPMA were used as the internal standard value and NIST SRM 610 was used as the external standard. NIST 612 and 614 glasses were utilised as secondary standards, and the concentrations of all analysed elements were within 10% of the standard values, with most within a <5% window.
Raman spectra of S rich and regular monazite-(Ce) were recorded at the University of São Paulo, using a LabRAM HR Evolution micro-Raman system working in back-scattering geometry, using a solid-state laser with a frequency of 633 nm and, equipped with 1800 gr/mm gratings, 1 cm–1 spectral resolution and a liquid-nitrogen-cooled CCD detector.
X-ray photoelectron spectroscopy depth profile measurements were conducted using ESCALAB 250xi XPS (Thermo Scientific, UK) with a MAGCIS™ Dual Beam Ion Source. Analysis was carried out using the selected area analysis mode with a nominal width of analysis of 200 μm and monochromated AlKα X-rays at 1486.6 eV. The MAGCIS source can generate both monatomic ion beams for profiling inorganic materials and also cluster ions for organic layer profiling. In this present case on depth profiling monazite-(Ce), the gun was selected in monoatomic mode with 3 keV ion energy and a raster size of 0.5 mm. The sputtering cycle was kept at 120 s each time. The charge neutraliser and X-ray source were only used during the acquisition of spectra, both being turned off during the sputtering cycle. Survey (wide) scans (step size 1 eV, pass energy 150 eV, dwell time 50 ms, number of scans 15) and narrow scans (step size 0.1 eV, pass energy 40 eV, dwell time 100 ms, number of scans 25) of the S 2p (binding energy, BE ≈ 164 eV), C 1s (BE ≈ 285 eV), O 1s (BE ≈ 531 eV), Ca 2p (BE ≈ 342 eV), P 2p (BE ≈ 132 eV), Y 3d (BE ≈ 153 eV), Ce 3d (BE ≈ 884 eV), La 3d (BE ≈ 835 eV), Nd 4d (BE ≈ 120 eV) and Fe 2p (BE ≈ 717 eV) regions were acquired. Data analysis were carried out using Thermo Avantage software version 5.952.
There is a possibility that redox reactions could be induced in the sample as it is sputtered with a mono-atomic argon beam for depth profiling. In order to fully investigate variation in the redox state of elements it was necessary to demonstrate that there was no redox interaction with the ion beam during surface cleaning. A single crystal of apatite from the Slyudjanka mica deposit (Voskoboinikova, Reference Voskoboinikova1938), Siberia, Russia, sampled at a depth of 120 m to avoid weathering effects (provided by J. Kynický) was rastered with the Ar+ beam over 31 cycles with narrow scan XPS collected after each cycle. There was no discernible chemical shift in Ca or P 2p electron binding energies (Supplementary Fig. S1A–B). A similar process was carried out on reagent grade FeSO4⋅nH2O over 10 raster cycles, again with no discernible chemical shift in the position of the S 2p electron binding energy peak (Supplementary Fig. 1C). From this we conclude that the Ar+ rastering process used for surface cleaning and depth profiling preserves robust redox state information in phosphates and for S species. Tests on homogenous areas of monazite-(Ce) suggest Ce3+ may oxidise to Ce4+ under the Ar+ beam, and so Ce narrow scans were analysed following polishing of the sample with no sputtering.
Results
Petrography
Silicified carbonatite samples are composed principally of amorphous silica phases, including opal and chalcedony, with minor hematite, calcite and quartz veins. The chalcedony contains abundant inclusions of iron-oxide minerals which and locally follow remnant cleavage planes, intersecting at 60° and 120°, after the original dolomite crystals (Fig. 3a). Small (25–50 μm) anhedral celestine grains are also distributed randomly within the amorphous silica. Both the iron and strontium from these minerals is likely to be sourced from the breakdown of the original dolomite, which is present in un-silicified samples and contains up to 10 wt.% FeO and 3 wt.% SrO (unpublished data, Broom-Fendley, 2019).
Fig. 3. BSE images (a–d, f) and EDS maps (e, g–j) of silicified carbonatite and monazite-(Ce) from Eureka. (a) Planes of hematite (Hem), intersecting at ~120°, cemented by chalcedony (Cdy) and local, anhdedral, celestite (Cls) grains. (b) Monazite-(Ce) (Mnz) with a large S-bearing monazite-(Ce) rim (S-Mnz), hosted in chalcedony. (c) Close-up of monazite-(Ce)/S-bearing monazite-(Ce) boundary, showing symplectic texture between calcite (Cal) and S-bearing monazite-(Ce). (d) Close-up of monazite and S-bearing monazite rods, showing pore formation at the monazite-(Ce)/S-bearing monazite-(Ce) boundary, with (e) showing changes in the S content of the S-bearing monazite over a small area. (f) Example of cross-cutting chalcedony and hematite veins, through monazite-(Ce) and S-bearing monazite-(Ce). (g–j) EDS maps demonstrating heterogeneous distribution of Ca and S in the S-bearing monazite (g–h), the presence of Ca (g) and Fe (i) inclusions in the S-bearing monazite assemblage, and the presence of chalcedony and hematite veins. Larger circles correspond to sites of XPS analysis, while smaller circles are areas of LA-ICPMS analysis. Numbering corresponds to analytical locations and data in Tables 2, S2, and Fig. 5.
Monazite-(Ce) in the silicified rocks is texturally similar to large monazite-(Ce) grains in un-silicified rocks. Common features include similar roundness, size (between ~0.1 and ~7 cm), orange colour, and the presence of hematite and rounded dolomite inclusions. However, monazite-(Ce) from the un-silicified samples has a distinct rim of S-bearing monazite-(Ce), up to 500 μm wide (e.g. Fig. 3b–h), and is also commonly cross-cut by later silica and hematite veins (Fig. 3f,i,j). The S-bearing monazite-(Ce) rim is porous and heterogeneous, with iron-oxide minerals (probably hematite) and calcite making up the rest of the assemblage; these non-phosphate minerals are complexly intermixed (Fig. 3c, f–j). At the margin of monazite-(Ce) grains, S-bearing monazite-(Ce) occurs as rounded, rod-like, globules which are interconnected by thin <5 μm veins, hosted in calcite, akin to a symplectic texture (Fig. 3c–e). The abundance of calcite intermixed with the S-bearing monazite-(Ce) decreases with increasing distance away from the core monazite-(Ce) grain, reaching a point where S-bearing monazite-(Ce) forms a seemingly homogeneous phase (Fig. 3g–h). Locally, S-bearing monazite-(Ce) is intermixed with Fe-oxide minerals (e.g. Fig. 3i).
Monazite-(Ce) composition
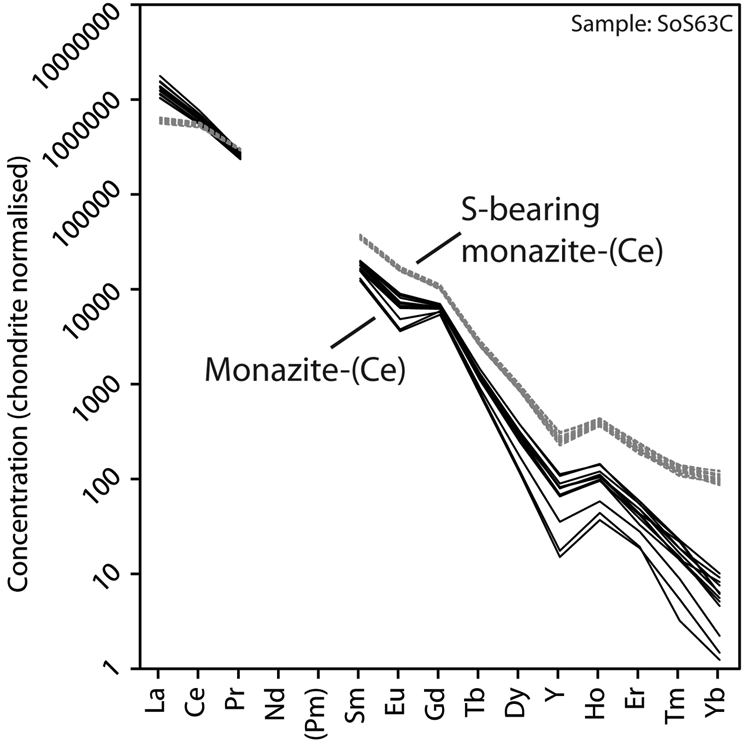
Representative EPMA and LA-ICPMS data for monazite-(Ce) and S-bearing monazite-(Ce) from Eureka are shown in Table 1, with the full dataset available in Supplementary Tables S1 and S2. Analysis locations are indicated on Fig. 3. The composition of monazite-(Ce) formed during carbonatite crystallisation is typical, in terms of its relatively low ThO2 and UO2 contents, for monazite-(Ce) from such rocks (cf. Chen et al., Reference Chen, Honghui, Bai and Jiang2017). However, chondrite-normalised REE contents exhibit a small negative Eu and slightly larger negative Y anomaly (Fig. 4), the former of which is atypical of carbonatite-derived minerals. S-bearing monazite-(Ce) has similar concentrations of ThO2 and SiO2 to the monazite-(Ce) grains, as well as the same Eu and Ce anomalies (Fig. 4). However, it has notably different SO3 (x̅ ≈ 9.6 wt.%), CaO (x̅ ≈ 6.6 wt.%), SrO (x̅ ≈ 4.0 wt.%), FeO (x̅ ≈ 1.0 wt.%), F (x̅ ≈ 0.8 wt.%) and Na2O (x̅ ≈ 0.2 wt.%) contents, well in excess of the EPMA detection limit (e.g. Fig. 3g–h). Trace elements such as U (x̅ ≈ 210 ppm), V (x̅ ≈ 79 ppm), Zr (x̅ ≈ 22 ppm), Nb (x̅ ≈ 10 ppm), Mo (x̅ ≈ 45 ppm) and Ba (x̅ ≈ 159 ppm) are also enriched relative to the monazite-(Ce) grains (all below detection except U, x̅ ≈ 80 ppm, Ba, x̅ ≈ 7 ppm). Moreover, S-bearing monazite-(Ce) is relatively HREE-enriched, and depleted in La and Ce contents, compared to grains which grew during carbonatite crystallisation.
Fig. 4. Chondrite-normalised REE distribution of monazite-(Ce) and S-bearing monazite-(Ce) from Eureka. Chondrite values after McDonough and Sun (Reference McDonough and Sun1995).
Table 1. Representative compositions of monazite-(Ce) and S-bearing monazite-(Ce) from Eureka (Sample SoS_63c).*
* Blank cells denote analyses below the EPMA detection limit; Y, Dy and U are below detection in all samples, Ba below detection in all but 2 S-bearing samples. Full dataset in Supplementary Table S1.
Table 2. Representative trace-element data (in μg/g) of monazite-(Ce) and S-bearing monazite-(Ce) from Eureka (Sample SoS_63c).*
* Blank cells denote analyses below the LA-ICPMS detection limit. Full dataset in Supplementary Table S2.
** 56Fe contents >15,000 μg/g may be contaminated with inclusions of an Fe-oxide mineral.
Oxidation state of sulfur
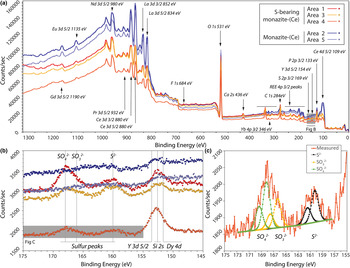
The locations of XPS analyses are shown in Fig. 3. Survey scans from binding energies of 0–1350 eV are shown in Fig. 5. The presence of S is identified clearly in the rim monazite-(Ce), as well as elevated HREE contents, Ca, and F (Fig. 5a,b). Narrow scans were obtained from the Y 3d peak as a proxy for the heavy rare earth elements (HREE; Fig. 5b), and sulfur peaks which vary between 160–170 eV, depending on their bonding environment. The peak position at 154 eV is characteristic of Y–O bonding in compounds (Mesarwi and Ignatiev, Reference Mesarwi and Ignatiev1993). Deconvolution of the S 2p spectra shows that S is present dominantly as SO42– (BE 168.14 eV; Yu et al., Reference Yu, Liu, Wang and Chen1990), but with a component of S2– (BE 160.25 eV; Vasquez, 1991) and SO32– (BE 166.34 eV; Abraham and Chaudhri, Reference Abraham and Chaudhri1986) (Fig. 5c).
Fig. 5. (a) X-ray photoelectron binding energy spectra of monazite-(Ce) from sites shown in Fig. 3. Note the HREE, S and Ca peaks present in the S-bearing monazite-(Ce). (b) Narrow scan of the Dy, Si, Y and S peaks. (c) Narrow scan of the S 2p binding energy peak indicating sulfur present as sulfite, sulfate and sulfide structurally bound in monazite-(Ce). Binding-energy peak positions from ThermoScientific, xpssimplified.com (accessed 2017), NIST XPS database (accessed 2017), Yu et al., (Reference Yu, Liu, Wang and Chen1990), Vasquez (Reference Vasquez1991) and Abraham and Chaudhri (Reference Abraham and Chaudhri1986).
Raman spectroscopy
The Raman spectra of S-rich and regular monazite-(Ce) is shown in Fig. 6. Band assignments of monazite-(Ce) have been proposed previously by Begun et al. (Reference Begun, Beall, Boatner and Gregor1981), Silva et al. (Reference Silva, Ayala, Guedes, Paschoal, Moreira, Loong and Boatner2006) and Lenz et al. (Reference Lenz, Nasdala, Talla, Hauzenberger, Seitz and Kolitsch2015). Tentative assignments of major Raman bands can be grouped into three different regions: 900–1100 cm–1 are attributable to the stretching of the PO4 tetrahedron; 450–700 cm–1 originate from bending vibrations within the PO4 group; and <450 cm–1 are related to lattice modes (Supplementary Table S3). No OH stretches were detected above 2900 cm–1 in the Raman spectra of the samples. However, the 633 nm laser excites an emission of Nd3+ (4f5/2 → 4l9/2 transition), which corresponds to bands in the range 3000–3600 cm–1.
Fig. 6. (a) Raman spectra of monazite-(Ce) and S-bearing monazite-(Ce) from sample SoS_63c. (b) Enlarged version of the above, demonstrating peak broadening and shifting.
The Raman spectra of S-rich and regular monazite-(Ce) are broadly analogous, but it is possible to observe some differences. In particular, between 900 and 1100 cm–1, there is an intense, sharp band at 968 cm–1, related to symmetric stretching, which has a shoulder at 991 cm–1 and a small band at 1056 cm–1, related to antisymmetric stretching for regular monazite-(Ce). In contrast the bands at 968, 991 and 1056 cm–1 are broadened significantly and shifted to 973, 1009 and 1074 cm–1 for S-rich monazite-(Ce) (Fig. 6b).
Discussion
Substitution of minor elements into monazite-(Ce)
Monazite-(Ce) is monoclinic (space group P21/n), comprising equal ratios of PO4 tetrahedra and REEO9 polyhedra (Ni et al., Reference Ni, Hughes and Mariano1995). The monazite-(Ce) structure differs from the tetragonal REE phosphate, xenotime-(Y), by a 2.2 Å offset along the [010] plane, allowing space for the larger LREE atoms, and leading to an additional REE–O bond compared with the 8-fold REEO site in xenotime (Ni et al., Reference Ni, Hughes and Mariano1995). In contrast to xenotime-(Y), the composition of monazite-(Ce) varies considerably in natural samples (e.g. Förster, Reference Förster1998), which might be a consequence of the irregular REE–O bond distances from the 9-fold coordination (Beall et al., Reference Beall, Boatner, Mullica and Milligan1981). In addition to isomorphous REE 3+ substitution, the 9 fold site also commonly accommodates other large cations, such as Th4+, U4+ and M2+ (e.g. Ca2+, Sr2+, Pb2+, Ba2+) with charge-balance achieved typically by the cheralite or huttonite substitution:
The above two mechanisms account for the incorporation of minor elements in monazite-(Ce) grains (without S) at Eureka. The similar number of Th4+ and Ca2+ + Sr2+ + Fe2+ apfu suggests that cheralite substitution predominantly accommodates the Th in this phase, with a minor huttonite component. A minor huttonite component can also predominantly account for Th4+ in the S-bearing monazite-(Ce), as the number of Si4+ apfu is equal, or greater than the Th4+ apfu in this phase.
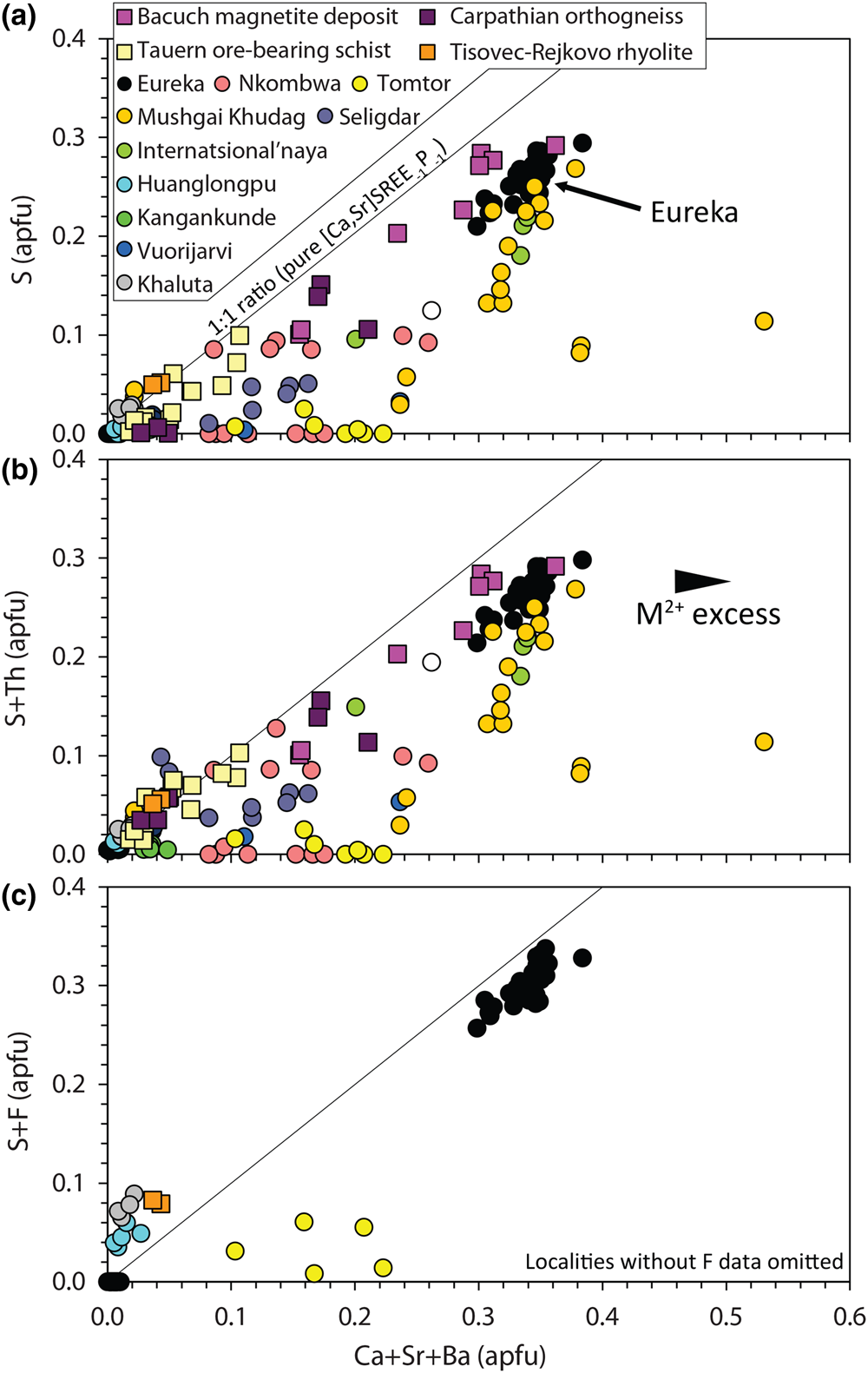
Sulfur substitution in monazite-(Ce) is relatively uncommon and, consequently, relatively understudied. Early work by Kukharenko et al. (Reference Kukharenko, Bulakh and Baklanova1961) suggested that S substitutes into the tetragonal PO4 site via a coupled substitution with anhydrite (also termed ‘clino-anhydrite’ and ‘anhydrite-celestite’ exchange by subsequent workers), based on a positive correlation between S and Ca + Sr cations:
An isomorphous substitution with Si has also been suggested (Williams et al., Reference Williams, Jercinovic and Hetherington2007):
Narrow XPS scans of the S peaks clearly demonstrate that sulfur is predominantly incorporated in monazite-(Ce) as sulfate (Fig. 5b–c). A positive correlation between Sr + Ca and S in the S-bearing monazite-(Ce) at Eureka is a strong indication that clino-anhydrite exchange (Fig. 7) accounts for the S content in this example, while low Si and Th contents suggest substitutions involving these elements do not play a role (Chakhmouradian and Mitchell, Reference Chakhmouradian and Mitchell1999; Bulakh et al., Reference Bulakh, Nesterov, Zaitsev, Pilipiuk, Wall and Kirillov2000; Wall, Reference Wall and Vladykin2004; Ondrejka et al., Reference Ondrejka, Uher, Pršek and Ozdín2007, Reference Ondrejka, Putiš, Uher, Schmiedt, Pukančík and Konečný2016; Pršek et al., Reference Pršek, Ondrejka, Bačík, Budzyń and Uher2010; Krenn et al., Reference Krenn, Putz, Finger and Paar2011; Laurent et al., Reference Laurent, Seydoux-Guilaume, Duchene, Bingen, Bosse and Datas2016). The capability of anhydrite to exist in the monoclinic crystal system, albeit at high pressures, gives credence to the possibility of clino-anhydrite substitution (Kahn, Reference Kahn1975; Borg and Smith, Reference Borg and Smith1975; Crichton et al., Reference Crichton, Parise, Antao and Grzechnik2005; Bradbury and Williams, Reference Bradbury and Williams2009). However, clino-anhydrite exchange cannot be the sole mechanism for the incorporation of sulfate, as it results invariably in an excess of M 2+ cations (Fig. 7a). This excess is not accommodated by cheralite substitution as there is insufficient Th4+ to balance the M 2+ excess (Fig. 7b). To accommodate this imbalance, Chakhmouradian and Mitchell (Reference Chakhmouradian and Mitchell1999) suggested the presence of an auxiliary substitution mechanism:
Fig. 7. Composition of monazite-(Ce) from Eureka compared with data compiled from other carbonatite complexes (circles; Kukharenko et al., Reference Kukharenko, Bulakh and Baklanova1961; Cressey et al., Reference Cressey, Wall and Cressey1999; Bulakh et al., Reference Bulakh, Nesterov, Zaitsev, Pilipiuk, Wall and Kirillov2000; Doroshkevich et al., Reference Doroshkevich, Ripp and Moore2001; Wall, Reference Wall and Vladykin2004; Lazareva et al., Reference Lazareva, Zhmodik, Dobretsov, Tolstov, Shcherbov, Karmanov, Gerasimov and Bryanskaya2015; Enkhbayar et al., Reference Enkhbayar, Sea, Choi, Lee and Batmunkh2016; Prokopyev et al., Reference Prokopyev, Doroshkevich, Ponomarchuk and Sergeev2017; Nikolenko et al., Reference Nikolenko, Redina, Doroshkevich, Prokpyv, Rafozin and Vladykin2018), the Internatsional'naya kimberlite (Chakhmouradian and Mitchell, Reference Chakhmouradian and Mitchell1999), and other published occurrences (squares) with SO3 >1% (Ondrejka et al., Reference Ondrejka, Uher, Pršek and Ozdín2007; Pršek et al., Reference Pršek, Ondrejka, Bačík, Budzyń and Uher2010; Krenn et al., Reference Krenn, Putz, Finger and Paar2011; Ondrejka et al., Reference Ondrejka, Putiš, Uher, Schmiedt, Pukančík and Konečný2016).
The absence of an OH peak in Raman data for the S-bearing monazite-(Ce) from Eureka (Fig. 6), however, means the above charge-balancing substitution is not a viable mechanism to accommodate the M 2+ excess. Moreover, our analytical totals from EPMA are within uncertainty of 100% (Table 1), rather than <100% as might be expected if OH substitution is taking place. We, therefore, propose that OH does not accommodate the M 2+ excess and, instead, we suggest an alternative auxiliary substitution where excess M 2+ cations are countered by F substituting for oxygen:
The additional charge-balancing of F considerably, although not entirely, reduces the M 2+ excess (Fig. 7c). The F content of monazite-(Ce) is not commonly analysed, but F concentrations up to 0.8 wt.% have been reported from Steenkampskraal monazite-(Ce) (Andreoli et al., Reference Andreoli, Smith, Watkeys, Moore, Ashwal and Hart1994). Moreover, F has been suggested to account for small excesses in Si after cheralite- and huttonite-type substitutions have been accounted for (Williams et al., Reference Williams, Jercinovic and Hetherington2007). Few data are available where both SO3 and F contents are reported and above the EPMA limit of detection, although monazite-(Ce) from the Tomtor, Khaluta and Huanglongpu carbonatites, and the Tisovec–Rejkovo rhyolite, are notable exceptions (Doroshkevich et al., Reference Doroshkevich, Ripp and Moore2001; Ondrejka et al., Reference Ondrejka, Uher, Pršek and Ozdín2007; Lazareva et al., Reference Lazareva, Zhmodik, Dobretsov, Tolstov, Shcherbov, Karmanov, Gerasimov and Bryanskaya2015; Chen et al., Reference Chen, Honghui, Bai and Jiang2017). In these instances, however, the proportion of Ca + Sr and S + F are not balanced, indicating that other mechanisms, such as co-substitution with SiO32– may be accommodating F (Williams et al., Reference Williams, Jercinovic and Hetherington2007). Nonetheless, in the absence of a better explanation to accommodate the M 2+ excess, we place considerable weight onto an auxiliary F substitution mechanism to accommodate charge imbalance.
Structural distortion in S-bearing monazite-(Ce)
The XPS binding energy spectra show that sulfur is present predominantly in S-bearing monazite-(Ce) as sulfate, as expected from the correlation between S and Ca + Sr contents. However, the X-ray photoelectron spectra also exhibits a minor additional peak for sulfide (S2–), and peak deconvolution indicates a small overlap with sulfite (S4+; Fig. 5c). Tests on iron sulfate show that sulfur does not undergo redox reaction under the Ar+ beam, and these peaks are significantly above background, so do not represent artefacts. The possibility of contamination of sulfite and sulfide from other phases is also unlikely as no other S-bearing phases are present in association with the S-bearing monazite-(Ce) (Fig. 3e, h). We cannot discount entirely the possibility that such phases may be present as nano-inclusions (e.g. Laurent et al., Reference Laurent, Seydoux-Guilaume, Duchene, Bingen, Bosse and Datas2016). However, 32S and 56Fe contents from LA-ICPMS analyses shows no clear correlation, indicating that pyrite is not present in the analyses (Supplementary Fig. S2; Fig. 3i–j).
Accommodating sulfur in monazite-(Ce) in oxidation states other than S6+ is somewhat challenging. In terms of charge-balance, S4+ could replace the Si4+ component in a pseudo-huttonite substitution, or as a coupled substitution with P5+:
The former option can be ruled out in this instance as Th4+ contents are very low, and can be accommodated by huttonite substitution. For sulfide, direct S2– for O2– may accommodate S2–:
Recent micro X-ray absorption near-edge structure analyses of both laboratory-synthesised and natural samples have indicated that sulfide and sulfite can occur in apatite (Konecke et al., Reference Konecke, Fiege, Simon, Parat and Stechern2017a, Reference Konecke, Fiege, Simon and Holtz2017b; Brounce et al., Reference Brounce, Boyce, McCubbin, Humphreys, Reppart, Stolper and Eiler2019; Sadove et al., Reference Sadove, Konecke, Fiege and Simon2019), which helps to support the concept that these species may occur in monazite-(Ce) as well. Ab initio studies of apatite support the mechanism for coupled substitution of S6+ and S4+ for P5+ (Kim et al., Reference Kim, Konecke, Fiege, Simon and Becker2017). Sulfite occurs as a trigonal pyramid with a lone electron pair replacing one of the O atoms in the substituted PO4 (or SO4). The stability of sulfite substitution in apatite is affected by the presence of the anion column, which repels the lone pair of electrons in the sulfite trigonal pyramid. The highest stability occurs in chlorapatite owing to increased distance between the Cl– anion and the electron pair, relative to OH– and F– (Kim et al., Reference Kim, Konecke, Fiege, Simon and Becker2017). The absence of the anion column in monazite-group minerals therefore suggests that sulfite substitution may be more stable than in apatite. Conversely, however, S2– is accommodated in the anion site in apatite (Kim et al., Reference Kim, Konecke, Fiege, Simon and Becker2017), and no such site is available in monazite-(Ce). Substituting S2– for O2– is difficult to reconcile with the monazite-(Ce) structure owing to the considerable size difference between these two anions. All three of the above substitutional mechanisms would result in mis-matched geometry, and would probably result in local defects and structural distortion. Moreover, these mechanisms also exacerbate the problem of M 2+ excess, as the charge-balancing contribution of S is reduced.
While we lack evidence to support conclusively the three substitution mechanisms suggested above, stretches in the Raman bands at 968, 991 and 1056 cm–1 do indicate changes in the P–O bond distance caused by structural distortion (Fig. 6). The sulfate ion is ~70% of the size of the phosphate ion (Shannon, Reference Shannon1976), suggesting the unit cell of monazite-(Ce) contracts as a result of increasing degrees of exchange with clino-anhydrite, which could potentially accommodate larger ions to compensate. Such a contraction is supported by the data of Bulakh et al. (Reference Bulakh, Nesterov, Zaitsev, Pilipiuk, Wall and Kirillov2000) who demonstrated that the unit cell volume of monazite-(Ce) with ~3% SO3 is ~2% lower than that of monazite-(Ce) with no S substitution. The smaller size of the sulfate ion, in comparison with the substituted phosphate, may also help explain the slightly elevated Mo and V contents, as both molybdate and vanadate ions form substantially larger tetrahedra than PO4. Ondrejka et al. (Reference Ondrejka, Uher, Pršek and Ozdín2007) noted elevated SO3 contents in As-bearing monazite-(Ce)/gasparite-(Ce) which, owing to the similar size of the arsenate, molybdate and vanadate ions, may be analogous. Similarly, we suggest that the slightly elevated contents of HREE and Zr are a consequence of this structural distortion, rather than reflecting any significant change in the formation environment or REE fractionation during transport.
Crystallisation environment of S-bearing monazite-(Ce) at Eureka
Sulfur-bearing monazite-(Ce) is relatively rare in carbonatites although this may, in-part, result from S not being included routinely in a monazite-(Ce) EPMA routine. A compilation of monazite-(Ce) compositions from different carbonatites was presented by Chen et al. (Reference Chen, Honghui, Bai and Jiang2017). The majority of these data have <1% SO3, in addition, published occurrences with monazite-(Ce) containing >1% SO3 are limited to Nkombwa, Zambia (Wall, Reference Wall and Vladykin2004); Vuoriyarvi and Seligdar, Russia (Kukharenko et al., Reference Kukharenko, Bulakh and Baklanova1961; Bulakh, Reference Bulakh, Nesterov, Zaitsev, Pilipiuk, Wall and Kirillov2000; Prokopyev et al., Reference Prokopyev, Doroshkevich, Ponomarchuk and Sergeev2017); and Mushgai Khudag, Mongolia (Enkhbayar et al., Reference Enkhbayar, Sea, Choi, Lee and Batmunkh2016; Nikolenko et al., Reference Nikolenko, Redina, Doroshkevich, Prokpyv, Rafozin and Vladykin2018). In all of these occurrences, monazite-(Ce) forms at a late stage in the crystallisation history of the carbonatites. Commonly, formation is considered to be related to a hydrothermal process, with temperature estimates from fluid-inclusion data between 385–315°C at Seligdar and 150–250°C at Mushgai Khudag (Prokopyev et al., Reference Prokopyev, Doroshkevich, Ponomarchuk and Sergeev2017; Nikolenko et al., Reference Nikolenko, Redina, Doroshkevich, Prokpyv, Rafozin and Vladykin2018).
There is little evidence for substantial hydrothermal alteration at Eureka. Local alteration includes small-scale quartz veins and carbonate recrystallisation. However, there is no evidence linking these events with the formation of the S-bearing monazite-(Ce). Instead, S-bearing monazite-(Ce) is considered here to be related to carbonatite weathering and duricrust formation. S-bearing monazite-(Ce) is found exclusively in silicified rocks (Fig. 3), which are restricted to ~5 m depth, and limited commonly to much shallower levels. Duricrusts are prevalent in the Namib Desert, with calcrete common in the area around Eureka (Viles and Goudie, Reference Viles and Goudie2013). Localised silcrete can form in calcrete owing to the pH-controlled inverse solubility relationship between silica and calcite, with precipitation of silica favoured when pH is <9, and vice-versa (Watts, Reference Watts1980). Mixing of Si-rich porefluids with alkaline water leads to Si precipitation, while Si-rich solutions in the presence of carbonate minerals become unstable in a saline or high CO2 environment (Watts, Reference Watts1980; Nash and Shaw, Reference Nash and Shaw1998). A change in salinity is possible in a continental environment by mixing meteoric water with saline lake fluids, while CO2 can be introduced into groundwater through biological activity or the decomposition of organic matter (Bustillo, Reference Bustillo2010). Alternatively, reduction of pH at oxic/anoxic boundaries, such as the through oxidation of H2S, can lead to silica precipitation (Bustillo, Reference Bustillo2010). Of these three possibilities, salinity change seems the most probable, owing to the close proximity of the Eureka carbonatites to ephemeral rivers and the salinity of local groundwater (encountered at ~60 m during drilling). However, when considering the S source, a contribution from H2S cannot be entirely excluded.
Formation mechanism for S-bearing monazite-(Ce) at Eureka
While the sole association of the S-bearing monazite-(Ce) with silicified rocks demonstrates that weathering and silicification play a key role in its formation, the exact growth mechanism of the S-bearing monazite-(Ce) is unclear. Solid-state diffusion is unlikely given the lack of a local source of S, and negligible diffusion rates in monazite below 800°C (Cherniak et al., Reference Cherniak, Watson, Grove and Harrison2004; Gardés et al., Reference Gardés, Jaoul, Montel, Seydoux-Guillaume and Wirth2006, Reference Gardés, Montel, Seydoux-Guillaume and Wirth2007). Overgrowths can also be excluded owing to the corroded and pitted texture of the core monazite-(Ce) grains (Fig. 3b,c). Instead, we propose that the S-bearing monazite-(Ce) probably formed via a dissolution–precipitation reaction (Putnis, Reference Putnis2002, Reference Putnis, Oelkers and Schott2009). Such a reaction involves fluid-mediated replacement of a phase to reduce the free energy of a system. The product phase(s) must be a lower molar volume or higher solubility in order for the reaction to propagate (Putnis and Ruiz-Agudo, Reference Putnis and Ruiz-Agudo2013). Although monazite is a relatively insoluble mineral (e.g. Poitrasson et al., Reference Poitrasson, Oelkers, Schott and Montel2004; Cetiner et al., Reference Cetiner, Wood and Gammons2005), dissolution–precipitation has been demonstrated experimentally across a wide range of pressures and temperatures (e.g. Hetherington et al., Reference Hetherington, Harlov and Budzyń2010; Harlov and Hetherington, Reference Harlov and Hetherington2010; Harlov et al., Reference Harlov, Wirth and Hetherington2011; Budzyń et al., Reference Budzyń, Harlov, Kozub-Budzyń and Majka2016). Texturally, the S-bearing monazite-(Ce) fits several characteristics of a dissolution–precipitation reaction (Putnis, Reference Putnis2002, Reference Putnis, Oelkers and Schott2009), such as: (1) the close relationship between the parent and product phases; (2) a sharp reaction front between these two phases; and (3) development of porosity and permeability in the product phase (Fig. 3d). Fractures also occur perpendicular to the reaction front (e.g. Fig. 3f), although these are localised and cross-cut the product phase, suggesting formation unrelated to the growth of S-bearing monazite-(Ce).
Although the textural evidence for a dissolution–precipitation formation mechanism is convincing, we note that no experimental work has yet demonstrated that such a process occurs under ambient conditions similar to those suggested to form the S-bearing monazite-(Ce) at Eureka. Indeed, the lowest-temperature dissolution–precipitation experiments on monazite-(Ce) demonstrate quite contrasting findings. Grand'Homme et al. (Reference Grand'Homme, Janots, Seydoux-Guillaume, Guillaume, Magnin, Hövelmann, Höschen and Boiron2018) showed that a relatively simple assemblage of monazite, quartz, H2O and NaOH dissolved at 300°C, but secondary monazite did not form. In contrast, Budzyń et al. (Reference Budzyń, Konečný and Kozub-Budzyń2015) found monazite does show dissolution and precipitation textures in an experiment at 250°C with a similar, but more complex, assemblage of monazite, K-feldspar, albite, muscovite, biotite, H2O and Na2Si2O5. At crystallisation temperatures below those of the above experiments, the more common LREE phosphate occurring in nature is the hydrated equivalent to monazite-(Ce), rhabdophane-(Ce) (Kolitsch and Holtstam, Reference Kolitsch and Holtstam2004). Nonetheless, rare occurrences of monazite-(Ce) in diagenetic, weathering and low-grade metamorphic environments have been described (e.g. Cooper et al., Reference Cooper, Basham and Smith1983; Oliveira and Imbernon, Reference Oliveira and Imbernon1998; Cabella et al., Reference Cabella, Lucchetti and Marescotti2001; Rasmussen and Muhling, Reference Rasmussen and Muhling2007; Čopjaková et al., Reference Čopjaková, Novák and Franců2011), indicating that monazite can form in such environments at low temperatures. It is not clear, however, why S-bearing monazite at Eureka is a more stable assemblage under such conditions, rather than a mixture of gypsum and monazite-(Ce).
Cross-cutting relationships indicate that chalcedony continued to form after the cessation of S-bearing monazite-(Ce) growth. Such growth would probably limit the permeability of the rock, reducing further ingress of S-bearing solutions. Complete silicification may, therefore, be a limiting factor in the alteration of the monazite-(Ce) to S-bearing monazite-(Ce).
Source of elements in the S-bearing monazite
Most of the elements in the S-bearing monazite are probably derived from the breakdown of local minerals. For instance, dissolution–precipitation textures of the core monazite-(Ce) grains indicate that these are the source for P and REE (Fig. 3b–e). Strontium may also be derived, in part, from the core monazite-(Ce) grains, but is probably derived predominantly from the release of Sr from the breakdown and silicification of dolomite. Dolomite breakdown is also the probable source of Ca. Owing to a lack of sulfide minerals in the carbonatite and surrounding Etusis schists and quartzites, breakdown of local sulfide minerals is a negligible source of sulfur for forming the S-bearing monazite-(Ce). An alternative source is the extensive gypcrete formations occurring along the coast of the Namib Desert, up to 70 km inland (Ekardt et al., Reference Ekardt, Drake, Goudie, White and Viles2001). The exact source of the S in the gypcrete is enigmatic, but possibilities include marine-derived, fog-borne H2S (Brüchert et al., Reference Brüchert, Currie and Peard2009) or dimethylsulfide (Ekardt and Spiro, Reference Ekardt and Spiro1999). While Eureka is located slightly further inland (~90 km from the coast) than the limit of gypcrete formation, wind-blown transport of gypsum has also been suggested as a source of sulfate for inland weathering (Ekardt et al., Reference Ekardt, Drake, Goudie, White and Viles2001). Sulfur isotope analyses would help further address this question, but are beyond the scope of the present study.
Conclusions and implications
Sulfur-bearing monazite-(Ce) occurs in silicified carbonatite at Eureka, Namibia, forming rims up to ~0.5 mm thick on earlier-formed monazite-(Ce) megacrysts. We present new XPS data which proves for the first time that the sulfur is predominantly accommodated as sulfate, via a clino-anhydrite-type coupled substitution mechanism. This largely confirms the assumption that S uptake into monazite-(Ce) is limited to oxidising conditions only, controlled by the oxidation state of the host rock (e.g. Laurent et al., Reference Laurent, Seydoux-Guilaume, Duchene, Bingen, Bosse and Datas2016). Nonetheless, minor sulfide and sulfite peaks in the X-ray photoelectron spectra suggest that more complex substitution mechanisms incorporating S2– and S4+ are possible and that a more cautious approach for interpreting crystallisation environment from the presence of S in monazite is warranted. Incorporation of S6+ through clino-anhydrite-type substitution invariably results in an excess of M 2+ cations, which previous workers have suggested is accommodated by auxiliary substitution of OH– for O2–. However, our new Raman data show no indication of OH–, and instead we suggest charge imbalance is accommodated through F– substituting for O2–. The accommodation of S in the monazite-(Ce) results in considerable structural distortion which can account for relatively high contents of ions with radii beyond those normally found in monazite-(Ce), such as the HREE, Mo, Zr and V.
In contrast to S-bearing monazite-(Ce) in other carbonatites, S-bearing monazite-(Ce) at Eureka formed through a prolonged weathering process. This is indicated predominantly by the localisation of the S-bearing monazite-(Ce) to silicified rocks which are constrained to the upper-most 5 m of exposure. Ca, REE and P were derived from the local dissolution monazite-(Ce) and dolomite, and S-bearing monazite formed via a coupled dissolution–precipitation mechanism. We suggest that S is sourced via protracted aeolian input, as is the case for local gypcrete formations. While large S-bearing monazite-(Ce) grains are likely to be rare in the geological record, formation of secondary S-bearing monazite-(Ce) in these conditions may be a feasible mineral for dating palaeo-weathering, especially weathered carbonatites. Co-substitution of PbSO4 during clino-anhydrite exchange is a notable challenge for dating S-bearing monazite-(Ce) (Krenn et al., Reference Krenn, Putz, Finger and Paar2011). Nonetheless, at Eureka the 208Pb concentration in the S-bearing monazite-(Ce) is approximately half that of the original grain, while U contents are higher.
Acknowledgements
Pete Siegfried (GeoAfrica) and Tim Smalley (E-Tech Metals) are both thanked for suggesting the importance of weathering at Eureka. Jindrich Kynický is thanked for supplying an apatite sample from Slyudjanka. The original sampling and fieldwork was funded by a Society of Economic Geologists Hugh McKinstry Fund award to SBF, with additional funding from the NERC SoS RARE consortium (NE/M011429/1; SBF, MS, MBA, DAB, DA and FW) and a NERC Industrial Innovation Fellowship (NE/R013403/1) to SBF. DA and MBA acknowledge the Brazilian research foundation FAPESP (process 2014/50819-9) for financial support. The authors are grateful for the thoughtful reviews of Martin Ondrejka, Callum J Hetherington and Jean-Marc Montel, as well as the editorial handling of Éimear Deady, Stuart Mills and Helen Kerbey.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2019.79









 Open access
Open access