



Stethoscopes are frequently used on multiple patients, and they have been implicated as vectors for nosocomial transfer of bacteria responsible for healthcare-associated infections (HAIs). It is well documented that practitioner stethoscopes are not routinely disinfected,Reference Holleck, Merchant, Lin and Gupta 1 , Reference Jenkins, Monash, Wu and Amin 2 and studies based on bacterial culture show that they may be contaminated with potential pathogens including methicillin-resistant and -sensitive Staphylococcus spp, multidrug-resistant P. aeruginosa, Acinetobacter spp, Enterococcus spp, Escherichia coli, Klebsiella spp, and Streptococcus spp.Reference Whittington, Whitlow, Hewson, Thomas and Brett 3 – Reference Tschopp, Schneider, Longtin, Renzi, Schrenzel and Pittet 5 Culture-based studies have also shown that thorough stethoscope decontamination can significantly reduce pathogen colony-forming units (CFUs),Reference Messina, Ceriale, Lenzi, Burgassi, Azzolini and Manzi 6 , Reference Raghubanshi, Sapkota, Adhikari, Dutta, Bhattarai and Bhandari 7 although the impact of actual practitioner practices is less clear. Culture-based studies are limited, however, because culture can only identify agents of a priori interest but not entire microbial communities that may be present.
In contrast to culture, which is focused on individual bacteria and is only semi-quantitative, emerging molecular approaches using next-generation sequencing can provide unbiased profiling of entire bacterial communities in a manner that is both comprehensive and highly quantitative.Reference Di Bella, Bao, Gloor, Burton and Reid 8 , Reference Hugerth and Andersson 9 These powerful approaches have revolutionized studies of the microbiome and of microbial ecology. Here, we used next-generation sequencing to investigate bacterial contamination on several types of stethoscopes in a medical intensive care unit (ICU), including stethoscopes carried by practitioners and used with multiple patients. We also investigated the effects of cleaning protocols that are used by practitioners in everyday practice.
Materials and Methods
Sample collection method (set A)
Stethoscope samples were collected in the medical ICU at the Hospital of the University of Pennsylvania. Stethoscope diaphragms were swabbed for 60 seconds using a flocked swab moistened with sterile saline (Copan Diagnostics, Murrieta, CA). Set A included swabs collected from 10 single-use disposable stethoscopes directly from the box prior to use (clean stethoscopes), 20 single-use disposable stethoscopes in-use in inpatient rooms (patient-room stethoscopes), and 20 stethoscopes being carried by physicians, nurses, and respiratory therapists (practitioner stethoscopes). Because low levels of microbial DNA are ubiquitous and bacterial DNA derived from collection instruments, reagents or the environment can confound microbiome studies,Reference Salter, Cox and Turek 10 , Reference Kim, Hofstaedter and Zhao 11 a set of background controls comprised of swabs moistened with saline (collected in parallel with the stethoscope sampling) were obtained on each collection date (n = 20). Swabs were stored at −80°C.
Standardized cleaning method (set B)
We sampled 10 additional ICU practitioner stethoscopes using the procedure described above, following which stethoscope diaphragms were cleaned with a hydrogen peroxide wipe (hydrogen peroxide 1.4%; Clorox Healthcare, Oakland, CA) for 60 seconds and allowed to dry. Stethoscopes were then swabbed again using the same protocol.
Practitioner preferred cleaning method (set C)
An additional 20 practitioner stethoscopes were collected. To ensure that the pre-cleaning swab would not affect the post-cleaning communities, the stethoscope diaphragm was sampled as described above, but only the left half of the diaphragm was swabbed. Stethoscopes were returned to the practitioner, and they were instructed to clean their stethoscopes using the method they usually would use to clean it between patients. Practitioners cleaned their stethoscopes with hydrogen peroxide wipes (n = 14), alcohol swabs (70% isopropyl alcohol; Coviden-Webcol, Mansfield, MA) (n = 3), or bleach wipes (sodium hypochlorite 0.55%; Clorox Healthcare) (n = 3), and duration of cleaning was determined by practitioner preference. Once the diaphragm was visibly dry, the right half of the diaphragm was then swabbed to capture post-cleaning bacterial communities.
Bacterial extraction
Swabs were cut directly into PowerSoil beadbeater tubes (MoBio, Quagen, Venlo, Netherlands). DNA was extracted using the PowerSoil DNA kit (MoBio), according to the manufacturer’s instructions except for an additional 10-minute, 95°C incubation step to improve DNA yield from hard-to-lyse bacteria. DNA was stored at −20°C.
Bacterial amplification sequencing and analysis
Extracted DNA was amplified in triplicate 25-μL reactions using barcode-labeled primers targeting the bacterial 16S rRNA gene variable regions 1 and 2 (V1V2), employing previously described polymerase chain reaction (PCR) primers and protocols.Reference Charlson, Diamond and Bittinger 12 , Reference Lauder, Roche and Sherrill-Mix 13 The PCR primers target sequences that are conserved among bacteria, whereas the region amplified is not conserved and provides sequence-based information for bacterial identification. Samples were purified using bead purification, quantified using PicoGreen (Fisher Scientific, Waltham, MA), normalized to 5 ng/μL per sample, pooled, and subjected to deep sequencing on the Illumina MiSeq platform. Clean swabs moistened in sterile saline and dry clean swabs were run in parallel as controls for instrument- or reagent-derived background sequences. Using the QIIME 1.91 pipeline,Reference Caporaso, Kuczynski and Stombaugh 14 sequences were clustered based on 97% similarity into de novo operational taxonomic units (OTUs), which serve as the basic taxonomic unit for subsequent analysis. To identify the bacteria, OTUs were aligned to the Greengenes reference database of bacterial sequences. Stethoscope set C was also amplified using primers targeting the 16S rRNA gene V4 regionReference Kozich, Westcott, Baxter, Highlander and Schloss 15 and was sequenced and analyzed using the same protocol. Sequences aligning with Streptophyta, which represent chloroplast DNA, were removed from the analysis.Reference Ubeda, Taur and Jenq 16
Bacterial DNA was quantified using 2 methods. First, the amount of amplification product generated by bar-coded PCR primer amplification during library preparation was used to estimate the relative amount of contamination in pre- and post-cleaning specimens, as previously described.Reference Bittinger, Charlson and Loy 17 In addition, 16S rRNA gene qPCR was carried out on a subset of samples using primers and protocols previously described.Reference Sherrill-Mix, McCormick and Lauder 18
To identify bacteria (taxa) that are commonly associated with HAIs, we specifically queried sequences from practitioner stethoscope samples of sets A and C for the presence of Staphylococcus, Pseudomonas, Acinetobacter, Clostridium, Enterococcus, Stenotrophomonas, and Burkholderia genera. The 16S rRNA gene sequences assigned to these genera in the QIIME pipeline were then manually aligned to the NCBI 16S rRNA sequence database with BLAST to confirm genus identity and, if possible, to generate species-level assignment. Because any 16S rRNA gene primer set may have unrecognized biases,Reference Tremblay, Singh and Fern 19 , Reference Fouhy, Clooney, Stanton, Claesson and Cotter 20 we did this using both V1V2 and V4 sequences of the 16S rRNA gene. Any sample with ≥10 sequence reads aligning to the genus was considered a positive hit.
Statistical analysis
Figures were generated and statistical tests carried out using R version 3.2.3 software (R Foundation for Statistical computing, Vienna, Austria). Richness (number of taxa) and alpha diversity (within-community measure that encompasses both richness and evenness) was carried out after sequence rarefaction to 1,000 reads.Reference Lozupone and Knight 21 Alpha diversity was calculated using the vegan package. The pairwise Wilcoxon rank-sum test was used to compare between-group differences for the alpha diversity analysis. We analyzed β diversity (a metric of between-community differences) was analyzed by unweighted and weighted UniFrac using the QIIME pipeline, and was used to perform principal coordinate analysis (PCoA).Reference Caporaso, Kuczynski and Stombaugh 14 , Reference Lozupone and Knight 22 The adonis function in the vegan package was used to test for statistical significance between groups of communities using PERMANOVA in the β diversity analyses. The Wilcoxon rank-sum test was used to examine the differences between groups of communities in quantification, richness and diversity, and the Student t test was used for the pre- and post-cleaning 16S quantification differences.
Results
Stethoscope microbiome community analysis
We first estimated total bacterial contamination of practitioner stethoscopes, patient-room stethoscopes, clean unused stethoscopes, and background controls in set A according to the quantity of 16S amplicon post-PCR amplification (Fig. 1(A)). Practitioner stethoscopes had significantly higher 16S amplicon concentration compared to both the patient-room and clean stethoscopes (P = .035 and P = .004, respectively; Wilcoxon rank-sum test). Patient-room stethoscopes had significantly higher concentrations than the clean stethoscopes (P = 1.8 × 10−5; Wilcoxon rank-sum test). Both practitioner and patient-room stethoscopes were significantly higher in 16S quantity than background controls, whereas clean stethoscopes were indistinguishable from the background controls (P = .967; Wilcoxon rank-sum test).
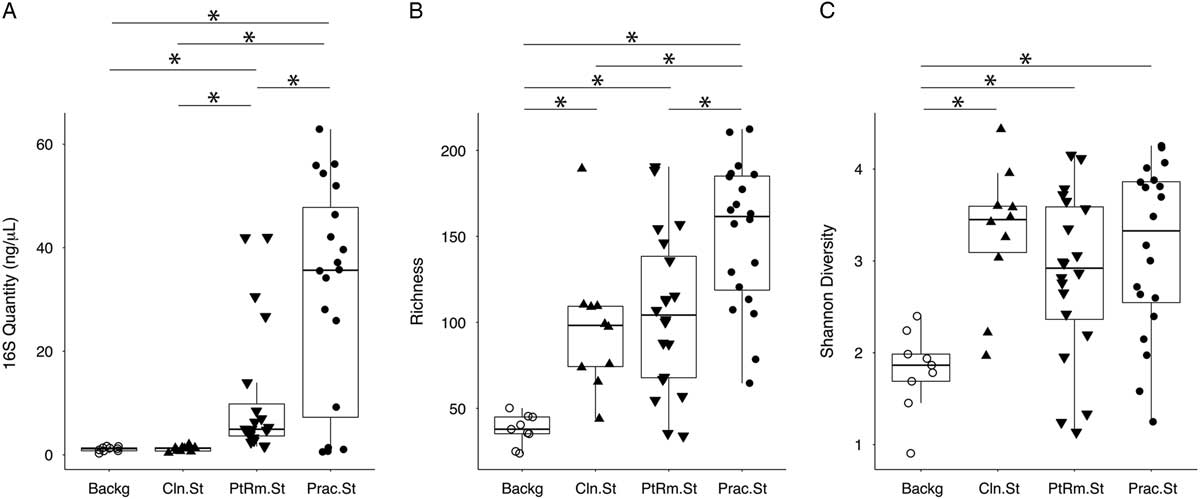
Fig. 1 Bacterial contamination, species richness, and α diversity of stethoscopes in the medical intensive care unit (ICU). Stethoscopes were analyzed for level of total bacterial contamination as quantified by 16S rRNA gene amplification (A). The nature of the communities were assessed by species richness, reflecting the number of different bacterial taxa identified (B), and α (within-community) diversity was calculated using the Shannon index, an indicator that encompasses both richness and evenness of distribution among the identified taxa (C). Prac.St, practitioner personal stethoscopes; PtRm.St, patient-room single-use stethoscopes; Cln.St, clean single-use stethoscopes; Backg, clean swabs that serve as controls for background bacterial DNA originating from swabs, saline, reagents or anywhere along the processing and sequencing pipeline. *P < .05 (Wilcoxon rank-sum test).
We analyzed species richness, which reflects the number of different taxa within each community (Fig. 1(B)). Practitioner stethoscopes had significantly greater richness than either patient-room or clean stethoscopes (P = .003 and P = .004, respectively; pairwise Wilcoxon rank-sum test). All stethoscope groups were significantly higher in richness than the background controls. There was no significant difference in richness between patient-room and clean stethoscopes.
We then assessed α diversity of the stethoscope bacterial communities using the Shannon diversity index, a metric that incorporates both richness and evenness of distribution, with higher diversity reflecting greater richness and more even distribution of taxa (Fig. 1(C)). Practitioner, patient-room, and clean stethoscopes were all significantly more diverse than the background controls (P = .0005, P = .003, and P = .0004, respectively; pairwise Wilcox rank-sum test). In contrast, there was no significant difference in Shannon diversity between stethoscope groups. Taxa identified on practitioner stethoscopes at >1% relative abundance within their respective communities are shown in a heatmap in Fig. S1.
To compare the overall bacterial communities of the different groups, we calculated UniFrac distances among samples using unweighted and weighted methods, then plotted them on principal coordinates analysis (PCoA) plots (Fig. 2). The UniFrac metric compares complex microbial communities based on the phylogenetic relatedness of the bacteria contained within the communities, and the PCoA plot provides an overview visualization of community relatedness.
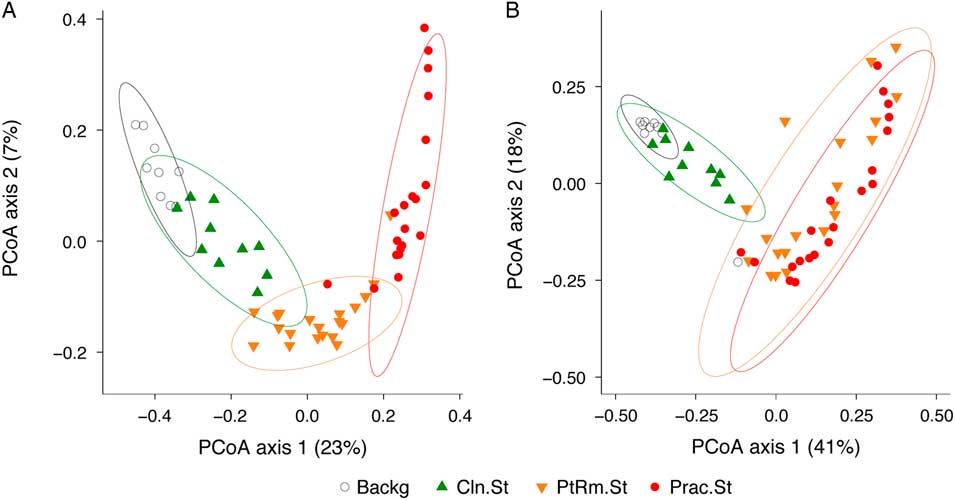
Fig. 2 Principal coordinates analysis (PCoA) of bacterial communities on stethoscopes in the medical intensive care unit (ICU). Bacterial communities were analyzed using unweighted (A) and weighted (B) UniFrac and were visualized by PCoA. UniFrac compares communities based on the phylogenetic relatedness of their constituent bacteria. Unweighted considers the presence or absence of bacteria, whereas the weighted approach also considers their abundance within each community. The PCoA provides a means of visualizing these relationships, and each symbol on the PCoA plot represents an individual bacterial community derived from one stethoscope or background control sample. All community types (practitioner stethoscopes, patient-room stethoscopes, clean stethoscopes and background controls) were significantly different by unweighted UniFrac (P < .05; PERMANOVA). By weighted UniFrac, the 2 in-use stethoscope types (practitioner and patient-room) did not differ from each other, nor did the 2 background samples (clean stethoscopes and background controls), but the 2 in-use stethoscopes were significantly different the 2 types of background samples (P < .05; PERMANOVA).
Using the unweighted UniFrac metric (Fig. 2(A)), which takes into account the presence or absence of taxa in different samples but not their relative abundance, bacterial communities were significantly different between all stethoscope groups (P < .001; PERMANOVA). Using the weighted UniFrac metric (Fig. 2(B)), which takes into account the presence or absence as well as relative abundance of bacteria comprising the communities, we observed that practitioner and patient-room stethoscopes differed from the clean stethoscopes and background controls (P < .001; PERMANOVA), but the practitioner and patient-room stethoscopes were not significantly different from one another (P = .106; PERMANOVA), nor were clean stethoscope and background controls (P = .07; PERMANOVA). These results suggest that both types of in-use stethoscopes differ substantially from the 2 types of control samples (clean stethoscopes and background), and that low-abundance taxa were mainly responsible for the differences between practitioner and patient-room stethoscopes, and for the differences between clean stethoscopes and background controls.
We next sought to determine which genera were responsible for the differences between bacterial communities on the in-use stethoscopes and the 2 types of controls on the weighted UniFrac PCoA. Figure 3 shows the top taxa responsible for separation of the communities plotted as vectors on the weighted PCoA, thus indicating which bacteria are responsible for differentiating communities located in distinct regions of the PCoA. The genera Methylobacterium, Pseudomonas, and Acinetobacter drove the separation of the clean stethoscopes and background controls from the practitioner and patient-room stethoscopes, indicating that these taxa are mainly derived from background sources in this sample set. Conversely, the practitioner and patient-room samples were characterized by Porphyromonas, Bacteroides, Granulicatella, Actinomyces, Prevotella, Streptococcus, Staphylococcus, Corynebacterium, and Propionibacterium, which are common oral, skin, and gut bacteria.

Fig. 3 Top 1% taxa that drive ordination on the weighted PCoA. To visualize the bacteria that are most responsible for differences between the groups of samples, taxa present at >1% abundance that are responsible for separation on the PCoA are shown, with the length of the vector proportional to its power to explain the separation. Practitioner and patient-room stethoscopes are distinguished by the presence of taxa such as Streptococcus, Staphylococcus, Propionibacterium, and other skin and gut flora, whereas clean stethoscopes and background samples are distinguished by detection of Methylobacterium, Pseudomonas, and Acinetobacter.
Effects of cleaning on stethoscope bacterial biomass and communities
Although patient-room stethoscopes are typically used only for a single patient, practitioner stethoscopes are used on multiple patients, raising the possibility of microbial transfer. Because practitioners may clean their stethoscopes between uses, we analyzed the effect of cleaning on the bacterial biomass on stethoscope diaphragms based on levels of 16S rRNA DNA. For 1 set of stethoscopes (set B; n = 10), we used a standardized cleaning method: vigorously wiping the diaphragm with a hydrogen-peroxide wipe for 60 seconds. For another set (set C; n = 20), we asked each practitioner to clean the diaphragm themselves with the method they usually use between patients. Bacterial contamination was quantified by the amount of 16S rRNA gene amplicon following barcoded PCR end-point amplification.
As shown in Fig. 4(A), standardized cleaning resulted in a significant decrease in stethoscope bacteria based on 16S rRNA gene amplification (P = 5.7 × 10−5; Student t test). Of the 10 practitioner stethoscopes, 5 dropped below the mean level seen on clean stethoscopes. In the practitioner-preferred cleaning method group (Fig. 4(B)), there was also a significant difference between pre- and post-cleaning measured by 16S rRNA quantification (P = .002, Student t test). However, only 2 of 20 stethoscopes dropped below the threshold of clean stethoscopes after practitioner cleaning. Similar results were obtained when bacterial quantification was done by 16S qPCR (Fig. S2).
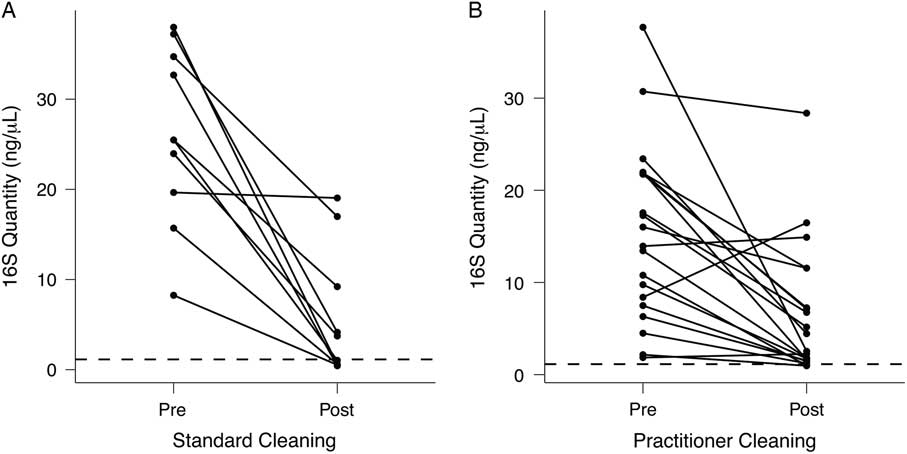
Fig. 4 Quantification of bacterial contamination on practitioner stethoscopes before and after cleaning. Bacterial contamination of practitioner stethoscopes was quantified based on the amount of amplicon following barcoded PCR amplification. Cleaning was done using a standardized (A) or practitioner-preferred method (B). The dashed line indicates the mean bacterial quantification measured on the clean stethoscopes. Both cleaning methods resulted in a significant reduction in bacterial contamination regardless of cleaning method (A: P = 5.69 × 10−5; B: P = .00174; Student t test). In the standardized cleaning group, 5 of 10 stethoscopes fell below the level of the clean stethoscopes as determined by amplicon concentration. In the practitioner-preferred cleaning group, 2 of 10 stethoscopes fell below the level of clean stethoscopes.
We then compared the bacterial community composition of practitioner Set C before and after practitioner cleaning (Fig. 5). In the unweighted Unifrac PCoA, which is equally impacted by high- and low-abundance taxa (Fig. 5(A)), the pre- and post-cleaning samples were significantly different from one another (P = .001; PERMANOVA) and were significantly different from the clean stethoscopes and background controls (P < .001; PERMANOVA). In contrast, no significance difference was observed between the pre- and post-practitioner cleaning in the weighted Unifrac PCoA, in which taxa are weighted based on their relative abundances (P = .274; PERMANOVA) (Fig. 5(B)). These results suggest that cleaning does not have a substantial effect on community structure and that low abundance (minority) taxa are mainly responsible for differences between the pre- and post-cleaning communities seen in the unweighted but not weighted analysis.
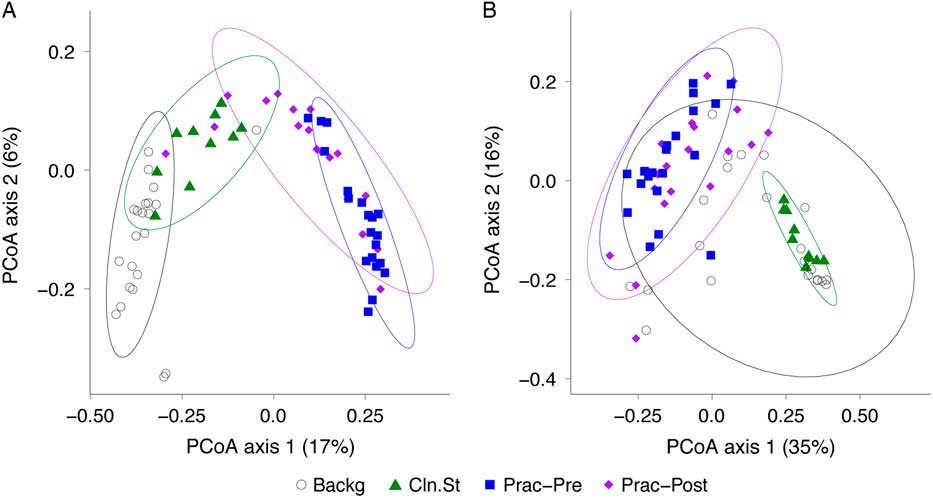
Fig. 5 PCoA of bacterial communities on ICU stethoscopes before and after practitioner cleaning. Stethoscopes bacterial communities before and after practitioner cleaning, along with clean stethoscopes and background controls, were analyzed using unweighted (A) and weighted (B) UniFrac and visualized by PCoA. All groups were significantly different in the unweighted UniFrac (P < .05; PERMANOVA), whereas by weighted UniFrac the pre- and post-cleaned stethoscopes types were not different.
Identification of nosocomial genera
We next investigated the presence of bacteria that are potential nosocomial pathogens. Samples from practitioner stethoscopes in sets A and C (prior to cleaning) were queried for relevant bacteria (Table 1). Both the V1V2 and V4 16S rRNA gene-variable regions were targeted to minimize the impact of potential primer biases.Reference Tremblay, Singh and Fern 19 , Reference Fouhy, Clooney, Stanton, Claesson and Cotter 20 In addition to aligning 16S rRNA gene sequences through the QIIME pipeline, sequences were manually aligned to databases by BLAST to optimize assignment.
Table 1 Identification of Potential Nosocomial Pathogens on Practitioner StethoscopesFootnote a

Note. SD, standard deviation.
a Sequence data from the practitioner stethoscopes in sets A and C were investigated for the presence of selected bacterial genera (g) that commonly cause hospital-acquired infections (HAIs), based on 16S rRNA gene V1V2 or V4 region sequences as described in the Methods section. Frequency indicates the proportion of practitioner stethoscopes with that genus. Relative abundance indicates the percentage of all bacterial sequences on a stethoscope that genus represents, across the practitioner stethoscope sets. For Staphylococcus, some sequences could be assigned at the species level (s), and those identified as S. aureus are indicated.
Sequences were identified from genera that are commonly associated with hospital-associated infections including Staphylococcus, Pseudomonas, Acinetobacter, Clostridium, Enterococcus, Stenotrophomonas, and Burkholderia. Most taxa could only be assigned at the genus level by both V1V2 and V4 16S rRNA gene sequences, although some Staphylococcus sequences could be identified at the species level and included S. aureus. All stethoscopes had Staphylococcus spp., and more than half of them were confirmed to carry S. aureus. Most stethoscopes also carried Pseudomonas and Acinetobacter. Approximately half of the stethoscopes had Enterococcus, Stenotrophomonas, and Clostridium, while Burkholderia was less frequent. The V4 results were overall highly concordant with V1V2 sequences, although the V4 region detected a somewhat higher prevalence of Burkholderia and Clostridium and somewhat less Stenotrophomonas.
For each of these genera, we calculated the relative abundance (ie, proportion of all bacterial sequences in a sample that were assigned to that genus) across all stethoscopes. Staphylococcus was present at 6.8%–14% relative abundance in the 2 practitioner stethoscope sets based on V1V2 and V4 sequences. Pseudomonas and Acinetobacter were less abundant and were present at slightly greater than 1% based on V4 sequences and ∼10-fold lower based on V1V2 sequences, whereas other genera were below 1% in abundance. Thus, practitioner stethoscopes that are used on multiple patients carry sequences of genera that include important nosocomial pathogens.
Discussion
This study is the first to apply comprehensive molecular profiling to understand stethoscope contamination in a healthcare setting. We found that stethoscopes carried by practitioners in an ICU, which are used on multiple patients, are significantly contaminated with a rich and diverse community of bacteria that includes genera associated with HAIs. We also examined the impact of cleaning and found a surprisingly modest effect on contaminating bacterial communities.
Taxa of skin, gut and oral sources dominated stethoscope bacterial communities, and genera associated with nosocomial infection were common. Staphylococcus was not only ubiquitous (found on 40 of 40 stethoscopes tested) but was also present at high abundance, representing 6.8%–14% of all bacterial sequences, depending on stethoscope set and target region queried. Most OTUs could not be assigned at the species level with either V1V2 or V4 sequences. However, definitive S. aureus assignment was possible for sequences on 24 of 40 practitioner stethoscopes. Therefore, at a minimum, more than half of these stethoscopes were contaminated by S. aureus. Pseudomonas and Acinetobacter were also widely present, with the exact frequency and relative abundance differing depending on the 16S variable region sequenced. Different 16S primer sets can have biases in the efficiency of detecting specific sequences,Reference Tremblay, Singh and Fern 19 , Reference Fouhy, Clooney, Stanton, Claesson and Cotter 20 and our findings using 2 different sets clearly establish that these taxa are widely present on stethoscopes, albeit at low relative abundances. Other nosocomial infection-related genera were also identified on practitioner stethoscopes at high frequency but lower relative abundances. Because this is the first study to analyze stethoscope contamination at the molecular level, it remains to be determined what amount of contamination is clinically relevant for potential nosocomial transmission.
We also applied these molecular methods to determine the impact of cleaning the stethoscope diaphragm, testing both a standardized approach and practitioners’ usual methods. Both methods significantly reduced the 16S DNA bacterial biomass but failed to bring contamination to the level of clean stethoscopes. The standardized cleaning method reduced more stethoscopes to the clean level (5 of 10) than the practitioner-preferred method (2 of 20). PCoA analysis of bacterial communities pre- and post-cleaning revealed a shift toward background communities detected by the analysis of community membership only, but not when bacterial relative abundance was incorporated (unweighted vs weighted UniFrac; Fig. 5). This results implicates low-abundance taxa as contributing the most to differences, whereas the more abundant taxa are not substantially altered by cleaning. Notably, although the CDC offers recommendations on stethoscope decontamination,Reference Rutala and Weber 23 studies suggest that it is infrequently practiced.Reference Holleck, Merchant, Lin and Gupta 1 , Reference Jenkins, Monash, Wu and Amin 2
Our study has several limitations. We were not able to identify bacterial taxa at the species level for most OTUs in our data set, and thus most HAI-associated bacteria could only be defined at the genus level. Future studies might complement comprehensive unbiased 16S rRNA gene analysis with species-targeted amplification. We were not able to identify drug-resistant species using this method. Because this is a DNA-based approach, it cannot distinguish between bacteria that are dead versus alive.Reference Emerson, Adams and Roman 24 Finally, we did not analyze fungal or viral sequences to understand the complete scope of organisms that stethoscopes carry in the ICU.
In summary, this study is the first comprehensive molecular analysis of bacterial contamination of stethoscopes in an ICU, the presence of potential nosocomial pathogens, and the impact of cleaning methods. Practitioner stethoscopes are contaminated by a plethora of bacteria, including organisms that may be associated with nosocomial infections. Cleaning reduces contamination but does not bring the bacterial biomass down to the level of clean stethoscopes nor does it significantly change the overall community composition. Thus, stethoscopes are a potential vector of HAI transfer. Useful future directions would be to use these molecular approaches to identify improved cleaning methods, enhance species-level identification of pathogens, quantify live versus dead bacteria, and define fungal and viral contaminants as well. In addition, shotgun metagenomic sequencing would be useful to analyze drug-resistance genes that might be carried between patients on practitioner stethoscopes.
Supplementary materials
To view supplementary material for this article, please visit https://doi.org/10.1017/ice.2018.319
Acknowledgments
We thank members of the ICU staff who assisted with this project and members of the Collman and Bushman labs who provided input and advice. We also thank D. Pegues for critical review of the manuscript.
Financial support
This work was supported by the National Institutes of Health (grant nos. R01-HL113252 and R61-HL137063). We received assistance from the Penn-CHOP Microbiome Program and the Penn Center for AIDS Research (grant no. P30-AI045008).
Conflicts of interest
The authors have no financial or other conflicts of interest relevant to this article.








