


INTRODUCTION
Creutzfeldt–Jakob disease (CJD) is the most common form of human prion disease. In CJD, normal prion proteins are converted to abnormal forms, which attain transmissibility, causing neuronal degeneration by accumulating in the central nervous system. Indeed, CJD is a lethal disease with a long incubation time, and once patients manifest clinical symptoms, they rapidly progress to serious disability and death.
CJD is categorized into four subtypes: sporadic, familial, iatrogenic, and variant. Sporadic CJD (sCJD) is the dominant form of CJD, the cause of which is unknown, and accounts for 85–95% of all CJD cases. Both familial CJD (fCJD), caused by a genetic mutation, and iatrogenic CJD (iCJD), caused by medical contamination, are rare, accounting for 5–15% and ⩽1%, respectively, of all CJD cases [Reference Masters1, Reference Ladogana2]. Variant CJD (vCJD) is acquired by ingestion of beef infected with spongiform encephalitis. Additionally, the incidence of sCJD is thought to be in the range of 0·5–1·5% per million people per year. In Japan, according to Nozaki et al. [Reference Nozaki3], sCJD, fCJD, iCJD and vCJD account for 77·2%, 16·7%, 6·1%, and 0·1%, respectively, of all CJD cases.
Previous studies have reported various aspects of the epidemiology and geographical variation of overall CJD incidence and mortality in Japan [Reference Arakawa4–Reference Doi9]. Nakamura et al. [Reference Nakamura7] reported standardized morbidity ratios (SMRs) for all CJD cases by prefecture from 1985 to 1996, and demonstrated that SMRs in Akita, Tochigi, and Tokyo were significantly higher than in the other prefectures [Reference Nakamura7]. Additionally, Nakamura et al. [Reference Nakamura8] reported SMRs for all CJD cases by prefecture from 1999 to 2004, and found higher SMRs in Akita and Yamanashi. In 2008, Doi et al. analysed the data on mortality of all CJD cases by municipality from 1995 to 2004, showing that areas around Shiga, Yamanashi and Akita had definite clusters of CJD cases [Reference Doi9]. Similarly, there have been a number of reports on the geographical variation of all CJD and sCJD incidence in other countries [Reference Matthews10–Reference Heinemann20]. In some of these studies, geographical clusters of CJD have been identified, with an impact of variations in the incidence of fCJD [Reference Raubertas21, Reference Belay22].
At present, sCJD is not thought to show regional clustering, partly because there is a lack of known plausible reasons. Some studies have claimed that apparent geographical clusters of all CJD may be caused by different incidences of fCJD [Reference Raubertas21, Reference Belay22], especially because fCJD is sometimes misdiagnosed as sCJD. Thus, the current study examined whether geographical clusters of sCJD exist, with sCJD and fCJD cases identified by genetic tests.
METHODS
Data
We extracted open access data, containing the annual number of new sCJD and fCJD cases by sex, age, and region (of residence) from 2001 to 2010 from annual reports of the Infectious Disease Incidence Trend Survey collected according to the Japanese Law Concerning the Prevention of Infections and Medical Care for Patients of Infections [23]. We also collected annual population data by sex, age and region during 2001–2010 (population estimates as of 1 August each year) and centroid of population in 2005 from the report of the National Census [24].
In the data, the diagnostic criteria of sCJD are stratified into three groups [25]. ‘Possible’ cases have the following criteria: progressive dementia; an atypical electroencephalogram (EEG) or no EEG performed; disease duration <2 years; and at least two of the following clinical features: myoclonus, visual or cerebellar disturbance, pyramidal/extrapyramidal dysfunction and/or akinetic mutism. ‘Probable’ cases (in the absence of an alternative diagnosis from routine investigation) are characterized as follows: have progressive dementia; have at least two of the following four clinical features: myoclonus, visual or cerebellar disturbance, pyramidal/extrapyramidal dysfunction, and akinetic mutism; have a typical EEG (generalized triphasic periodic complexes at about 1/s) whatever the clinical duration of the disease; and/or a positive 14–3–3 assay of cerebrospinal fluid; and a clinical duration leading to death in <2 years. ‘Definite’ cases are as follows: have neuropathological confirmation; and/or have confirmation of protease-resistant prion protein (immunocytochemistry or Western blot); and/or have scrapie-associated fibrils. In the current study, ‘definite’, ‘possible’ and ‘probable’ cases were regarded as sCJD.
The study conforms to the ethical guidelines for epidemiological research issued by the Ministry of Education, Culture, Sports, Science and Technology and the Ministry of Health, Labour and Welfare in Japan. Because the data in this study have no identifiable personal information, institutional review board approval was not required.
Statistical analysis
To investigate the impact of age on sCJD incidence, our data were divided into 15 age groups; the first age group was 0–4 years, with subsequent groups at 5-year intervals up to the >70 years age group.
Annual incidence rates (per 1 000 000 population) were calculated using newly diagnosed cases as the numerator and age- and sex-specific estimates of the population as the denominator. Age-and sex-specific incidence rates were calculated for the overall study period and for each calendar year from 2001 to 2010; confidence interval (CI) estimates were calculated as 95% CI estimates of the number of subjects (Poisson distribution) divided by the population in each year.
We performed Poisson regression analysis to investigate the sex differences by adjusting age intervals for sCJD incidence, and to investigate the differences in years or periods for sCJD and fCJD incidence, respectively.
To analyse the geographical distribution of the incidence of sCJD in each prefecture of residence, the ratio of standardized incidence (SI) was calculated, as defined by the actual number of new patients divided by the expected number of new patients. The expected number of new patients was calculated using the incidence for the whole of Japan and the population in each prefecture. With the assumption that the incidence of sCJD by prefecture followed a Poisson distribution, the SI ratios were estimated using Poisson regression. For spatial disease clustering, the Kulldorff method was used [Reference Kulldorff26]. For the statistical analysis, SAS version 9.1 (SAS Institute Inc., USA) and R version 3.0.0 (R Foundation, Austria) were used.
RESULTS
Descriptive epidemiology of sCJD
Based on the data from 2001 to 2010, the incidence of sCJD was 1·026 per million/year (637 cases) for men and 1·132 per million/year (733 cases) for women, with men accounting for 46·6% of all cases. The incidence was close to 0 in the group aged <40 years, and it exponentially increased with age in individuals aged >45 years both in men and women (Fig. 1). The incidence of sCJD was higher in men than in women for those aged >60 years (P = 0·001), although the number of sCJD cases was greater in women in the oldest age group.
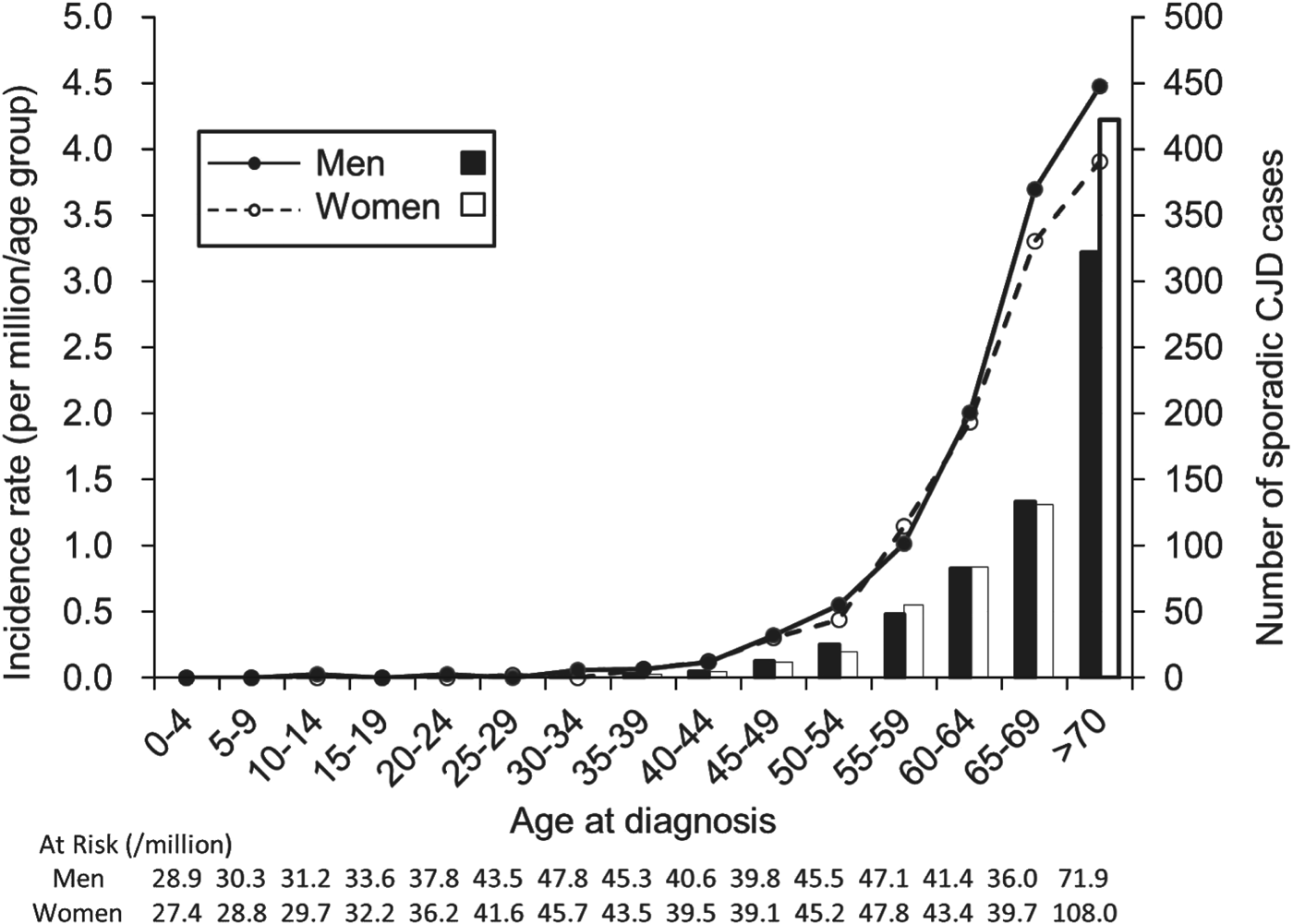
Fig. 1. Incidence and number of sporadic Creutzfeldt–Jakob disease (CJD) cases by sex in each age group at diagnosis for men (–●–; ■) and women (–○–, □).
Temporal trend in sCJD incidence and comparison with fCJD incidence
During 2001–2010, the incidence of sCJD in men varied from 0·837 to 1·321 per million/year, and that in women varied from 0·882 to 1·375 per million/year (Fig. 2). There was no significant increase in sCJD incidence in either sex during this period (P = 0·290). After 2006, although the incidence appeared to be slightly higher in women, there was no significant difference.
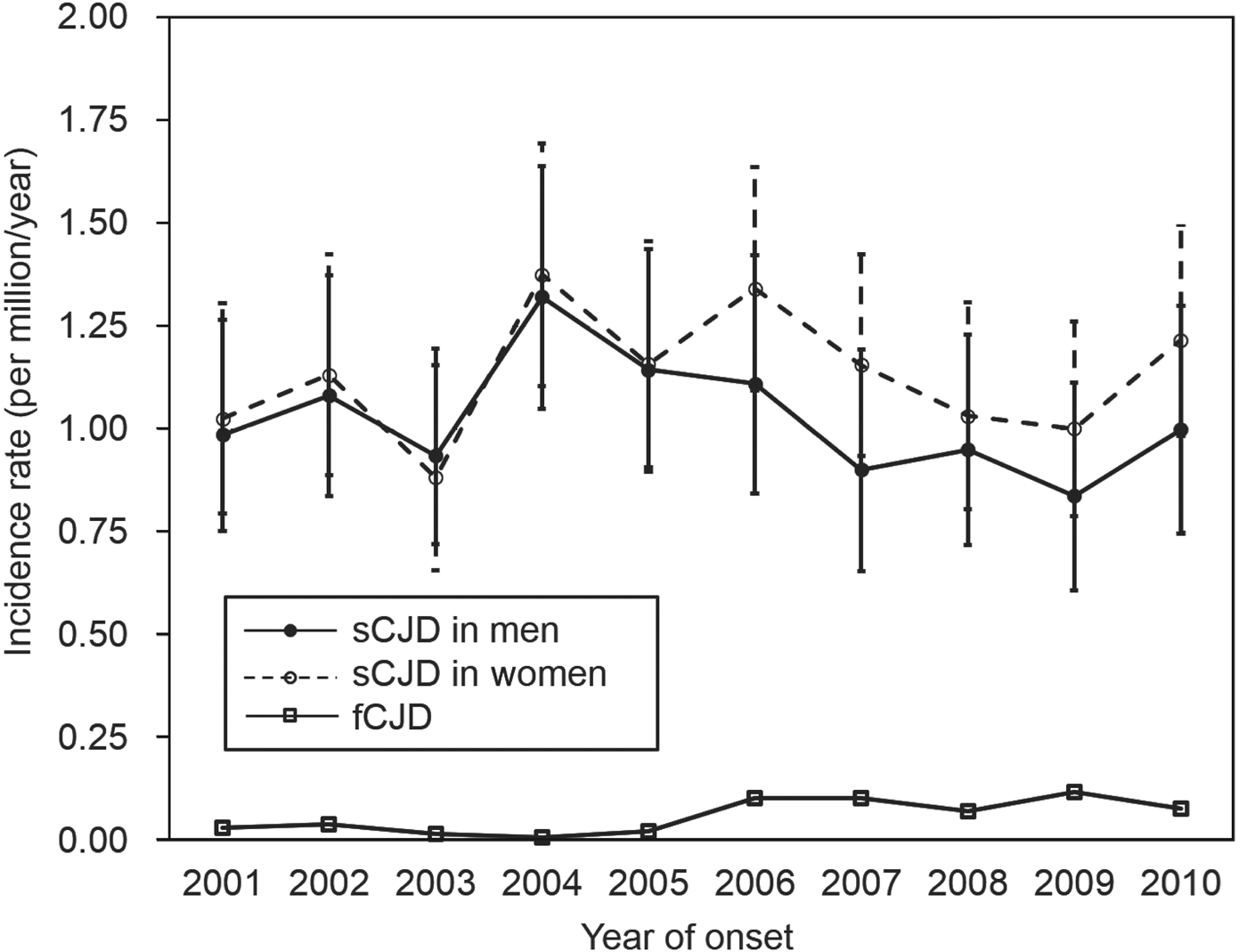
Fig. 2. Incidence (with 95% confidence intervals) of sporadic Creutzfeldt–Jakob disease (sCJD) and familial Creutzfeldt–Jakob disease (fCJD) by sex per year.
When comparing sCJD and fCJD, the incidence of fCJD (0·059 per million, 75 cases) was much lower than that of sCJD (1·080 per million, 1370 cases). Moreover, fCJD incidence between 2006 and 2010 (range 0·0708–0·1180) tended to be higher than that between 2001 and 2005 (range 0·0079–0·0395) (P < 0·001), indicating an apparent increase in incidence of fCJD in the latter period when nationwide genetic tests were introduced. At the same time, the ratio of fCJD to sCJD increased from 0·021 (15:699) in 2001–2005, to 0·089 (60:671) in 2006–2010.
Geographical distribution of sCJD
Figure 3 illustrates the geographical distribution of sCJD in Japan, as represented by age-adjusted SI obtained for the period 2006–2010. Data for 2001–2005 were not included, because they were likely to be less accurate than data for 2006–2010, when diagnosis was confirmed by genetic tests. The age-adjusted SI for 2006–2010 was in the range 0·387–1·879, implying a 4- to 5-fold difference between the lowest and highest prefecture. The highest scores of >1·5 (or >1·5 times higher than the mean incidence) were found in Ishikawa (1·879), Shimane (1·860), Yamanashi (1·835), Ehime (1·695), Saga (1·644), Yamaguchi (1·608), and Kochi (1·584). Based on spatial disease clustering, Ishikawa and the area from Kochi to Ehime appeared to be the site of two major clusters of sCJD (P = 0·028 and P = 0·042, respectively).

Fig. 3. Geographical distribution by age- and sex-adjusted standardized incidence (SI) ratios of sporadic Creutzfeldt–Jakob disease during 2006–2010. The SI ratio of each prefecture is categorized according to intervals of 0·5.
DISCUSSION
The incidence of sCJD was markedly associated with age, which is similar to the finding in previous studies on all CJD cases [Reference Gubbels27–Reference Doi29]. Regardless of whether sCJD is a non-infectious or infectious disease, the incidence exponentially increased with age. If sCJD is a non-infectious disease, spontaneous intrinsic production and proliferation of abnormal prions would contribute to the age-dependent increase in its incidence. If sCJD is an infectious disease, opportunistic infection of prions could contribute to the marked increase in older frail individuals. In either case, the development of drugs to block the transformation process from normal to abnormal prions would be the key for preventing sCJD. Additionally, we found a higher incidence of sCJD in men than in women for individuals aged >60 years, which is compatible with previous studies [Reference Nakamura7, Reference Gubbels27]. However, this result needs to be interpreted with caution because increased survival of healthy older women can dilute the incidence of sCJD in such age groups. Indeed, despite the greater number of women with sCJD in the oldest age group (>70 years), the incidence of this age group was lower than in men, due to the existence of more women without sCJD.
The annual incidence of sCJD found in this study (men: 0·837–1·321 per million; women: 0·882–1·375 per million) appears to be higher than that reported by Nozaki et al. (0·55–0·87 per million) for 1999–2008 in Japan [Reference Nozaki3], whereas it is similar to a report from Germany (0·8–1·6 per million) and the mortality rate from Australia and Canada (1·39 per million) [Reference Ladogana2, Reference Heinemann20]. The difference between this study and that of Nozaki et al. may be related to the data sources. Thus, our data were based on annual reports in the Infection Disease Incidence Trend Survey (1370 cases over 10 years) and the data of Nozaki et al. were based on a prospective cohort by the CJD Surveillance Committee in Japan (1019 cases over 10 years). Additionally, we found that the incidence ratio of fCJD to sCJD was higher in 2006–2010 than in 2001–2005, which was probably a result of the improved diagnosis of fCJD after the nationwide introduction of genetic tests in 2006.
The clusters of higher sCJD incidence, as identified in this study, differed from those in previous reports. Compared to the studies from 1999 to 2004 and from 1995 to 2004 [Reference Nakamura8, Reference Doi9], Ishikawa and the area from Ehime to Kochi were newly identified as having sCJD clusters. Although Yamanashi had a cluster of total CJD in the two previous studies [Reference Nakamura8, Reference Doi9], it was not the case in this study, although the age-adjusted SI (>1·5) was relatively high for the area. This could be related to the differences in data sources, categorization of disease, statistical methods used, definition of geographical areas, definition of clusters, and study duration, and thus the epidemiological implication is not clear. Other potential reasons are that the previous nationwide surveys in Japan included all CJD patients and used mortality rates, whereas the current study included only sCJD (excluding fCJD) [Reference Nakamura7–Reference Doi9], and that the period of data collection (2001–2010) was different from that of previous studies [Reference Nakamura7–Reference Doi9].
Of note, even when cases of sCJD and fCJD were separated, certain geographical clusters of sCJD remained, suggesting region-specific factors in the clusters, such as hereditary background or other local factors. Specifically, the geographical clusters were scattered in the western half of Japan, and whether there are any common underlying factors remains to be explored. Additionally, although our findings are based on data by prefecture, investigation of smaller areas, such as cities, could help elucidate further the aspect of geographical clustering, and potentially contribute to the clarification of sCJD aetiology.
When interpreting the results of the current study, certain limitations need to be addressed. First, because the numbers of CJD cases were relatively small, we used statistical methods appropriate for such situations. The rare occurrences of a disease generally follow a Poisson distribution, and we assumed that the number of subjects in each prefecture was independent and identically distributed with a Poisson distribution. The expected number of cases (as denominator) for the estimated SI ratio would be robust, because the incidence in all Japan and the population in each prefecture are stable. Furthermore, the variance of the SI ratio was not wide, as determined from 95% CIs (data not shown) and the value of McFadden's R 2, as goodness of fit of the Poisson regression model was 0·62. Second, because the autopsy rate is low in Japan [Reference Nozaki3] and mortality data were not available, our data are based on clinical diagnoses, allowing for error from misdiagnosis. Indeed, other diseases such as rapidly progressive Alzheimer's disease can be misdiagnosed as CJD. For example, a recent study in the USA examined 1106 cases diagnosed with CJD, and found 352 (32%) cases were prion free [Reference Chitravas30]. Despite such limitations, the findings of this study help improve the accuracy of CJD epidemiology as CJD cases were confirmed by genetic tests which separated sCJD from fCJD.
In conclusion, based on the data obtained between 2006 and 2010, when CJD diagnosis was confirmed with genetic tests, certain geographical clusters of sCJD were identified, suggesting the existence of underlying region-specific factors affecting the incidence of the disease in Japan. Further investigation of geographical clustering may aid in the elucidation of the aetiology of sCJD.
ACKNOWLEDGMENTS
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
DECLARATION OF INTEREST
None.



