Differential susceptibility theory predicts that some individuals are more susceptible to environmental influences than others, not only for the worse but also for the better (Belsky, Bakermans-Kranenburg, & van IJzendoorn, Reference Belsky, Bakermans-Kranenburg and van IJzendoorn2007). Genetic differential susceptibility suggests that specific genotypes previously considered “risk genes” are markers of susceptibility (Bakermans-Kranenburg, van IJzendoorn, Pijlman, Mesman, & Juffer, Reference Bakermans-Kranenburg, van IJzendoorn, Pijlman, Mesman and Juffer2008; Belsky et al., Reference Belsky, Jonassaint, Pluess, Stanton, Brummet and Williams2009). This implies a bold set of hypotheses fundamentally different from the conventional idea of diathesis stress that only in adverse circumstances do vulnerable individuals show deviant developmental pathways compared to their more resilient peers (Belsky & Pluess, Reference Belsky and Pluess2013). Differential susceptibility proposes that the so-called vulnerable carriers of risk genes will outperform their peers in supportive environments, demonstrating their susceptibility instead of their vulnerability.
A bold theory requires audacious tests and thorough empirical scrutiny. Randomized control trials (RCTs) are the most stringent experimental test in the study of human development. The quantitative combination and analysis of the available experimental evidence with meta-analytic methods goes beyond the sometimes limited power of individual experiments to provide a more definite proof or refutation of differential susceptibility. In this paper, we aim to test differential susceptibility theory by examining the RCTs on genetic differential susceptibility conducted to date (i.e., March 2014), including the studies reported in the Special Section of Development and Psychopathology (January 2015) on genetically moderated interventions.
Correlational Gene × Environment (G × E) Susceptibility Studies
Genetic differential susceptibility studies constitute a subclass of G × E studies, with the additional directed hypothesis of a specific type of crossover interaction to be found in environments with a sufficiently broad range from positive to negative characteristics, and developmental or behavioral assessments that not only cover negative consequences but also positive outcomes (for reviews, see Belsky & Pluess, Reference Belsky and Pluess2009, Reference Belsky and Pluess2013). Until recently, most G × E studies testing differential susceptibility have used correlational designs. The first G × E study testing the moderating effect of the genotype in a for better and for worse manner showed that children carrying the dopamine receptor D4 gene (DRD4) seven-repeat allele displayed the most externalizing behavior at 39 months when their mothers were observed to be insensitive during home observations at 10 months of age but the least externalizing behavior when their mothers were highly sensitive (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2006). Five years later, the number of studies focusing on dopamine-system related genotypes had accumulated sufficiently to meta-analyze the data (k = 15, N = 1,232). The meta-analysis confirmed that genotypes that in adverse contexts put children at risk for behavior problems made them also benefit more from support (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2011).
A larger number of correlational G × E studies have been conducted using serotonin-system related genotypes, in particular the serotonin transporter linked polymorphic repeat (5-HTTLPR), as a marker of differential susceptibility (k = 77, N = 9,361; van IJzendoorn, Belsky, & Bakermans-Kranenburg, Reference van IJzendoorn, Belsky and Bakermans-Kranenburg2012). In the total study set, including studies with non-Caucasian and mixed ethnicities, children with short serotonin transporter (5-HTT) alleles were more negatively affected by adverse contexts than carriers of two long alleles with regard to negative outcomes, but they did not benefit significantly more from positive environments. This result seemed to support the diathesis–stress model that short alleles should be considered “risk” alleles only making individuals more vulnerable to environmental adversity but not more open to supportive contexts (van IJzendoorn et al., Reference van IJzendoorn, Belsky and Bakermans-Kranenburg2012). Because ethnicity was a significant moderator of the effect sizes, the analyses were repeated for studies with Caucasian participants (k = 52, N = 6,626). In this set, clear evidence for differential susceptibility emerged. Carriers of short alleles appeared to be more sensitive to negative as well as positive environmental influences than individuals homozygous for the long allele. Ethnicity might thus be an important moderator of genetic differential susceptibility.
G × E Experiments
In addition to the more common correlational studies, a growing body of work concerns experimental G × E studies. Genetic differential susceptibility experiments are experimental G × E studies testing the “bright side” of the moderating role of genotypes that have been shown to be related to vulnerability to negative conditions (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2015). From the perspective of diathesis–stress theory, “vulnerable” or “risk” genotypes are associated with bad outcomes when exposed to stressful or unfavorable environments. Differential susceptibility theory predicts that carriers of the very same genotypes conferring risk in negative circumstances profit most from interventions aimed at changing the (rearing) environment for the better.
The most decisive G × E experiments (G × Experimental E [G × eE]) are RCTs fulfilling the requirement of randomized assignment of participants to control group and intervention, that is, to a putative improvement of the environment. Genetic variation can be a fixed factor but randomized environment is a necessary condition of experimental G × E (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2015). Experimental G × E studies provide causal, not just observational and correlational, evidence that existing interventions vary in their efficacy as a function of the genetic makeup of the individuals exposed to such interventions. This is a necessary next step in the differential susceptibility research program to overcome the limitations of correlational research on differential susceptibility (van IJzendoorn et al., Reference van IJzendoorn, Bakermans-Kranenburg, Belsky, Beach, Brody and Dodge2011).
Differential susceptibility experiments have at least three distinct advantages compared to correlational genetic differential susceptibility studies. First, genes and the environment are uncorrelated. In RCTs, the environment is manipulated in standard ways, and randomization breaks the potential gene–environment correlation (rGE). Genetic factors influencing an individual's exposure to particular environments could make those environments themselves heritable (Jaffee & Price, Reference Jaffee and Price2007). Correlations between genotype and environment cannot play a contaminating role in experimental interventions changing the environment, however, because genes may only moderate the effectiveness of the intervention. Of course, in correlational G × E studies, the rGE is often set aside by ascertaining that the genetic marker is not correlated with the indicator of the environment, but this is not a definite proof of the absence of rGE because some unmeasured genetic component might be responsible for the environment. In behavioral genetic studies, environmental measures themselves have been shown to be partly heritable (Jaffee & Price, Reference Jaffee and Price2007), and in particular, self-report assessments of the environment might be liable to heritable response biases (Eaves et al., Reference Eaves, Heath, Martin, Maes, Neale and Kendler1999). Only random assignment creates true independence of (change in) the environment and genetic makeup (van IJzendoorn et al., Reference van IJzendoorn, Bakermans-Kranenburg, Belsky, Beach, Brody and Dodge2011).
Second, G × E experiments avoid or decrease the risk of unequal measurement errors in the G × E equation, that is, the varying error components in the interaction equation of genetics and environment. If genetic assessments are done in a careful way but broad or “quick and dirty” measures are used for the environment (e.g., self-reported retrospective childhood experiences), the error components are smaller for genes than for the environment, creating risks for Type 1 and Type 2 errors. G × E findings are critically dependent on accurate assessments of both the genotype and the environment (McGuffin, Alsabban, & Uher, Reference McGuffin, Alsabban and Uher2011). A huge strength of experiments with a well-defined, standardized manipulation of a specific dimension of the environment is the reduction of measurement error in the environment. Of course, ineffective interventions not resulting in measurable change of the environment do not contribute to a reduction of measurement error in the environment. Assessing the changes in the environment might be important to check the impact of the manipulation and to examine dose–response relations between environmental change and outcome in the experimental condition (for an example, see Bakermans-Kranenburg et al., Reference Bakermans-Kranenburg, van IJzendoorn, Pijlman, Mesman and Juffer2008). Even when the experimental intervention is effective in creating a different environment for the experimental group compared to the control subjects, it is still crucial to assess the outcome of the manipulation in a sensitive and reliable way.
Third, randomized G × E experiments provide substantially increased statistical power compared to correlational G × E studies. Experimental studies create more variance in the product term because interventions make experimental participants maximally different from controls. Correlational studies generally show truncated distributions at the extremes and many observations toward the center of the distributions. This is because of unavoidable selective recruitment and attrition especially in the eccentric parts of the distribution. As a result, the power of experimental G × E studies may be more than 10 times larger than that of correlational studies. In simulations with two factors (e.g., treatment and genotype) McClelland and Judd (Reference McClelland and Judd1993) demonstrate that, independent of the effect size of the moderator, 1,300 subjects are needed to achieve the same power in a correlational study as in an experiment with 100 subjects. They show that this does not depend on the effect size of the moderator (McClelland & Judd, Reference McClelland and Judd1993).
Why Meta-Analysis?
Some G × E experiments seem to support genetic moderation of manipulation of the environment for the better, whereas others only show a main effect of the intervention. In narrative reviews of the literature, it is rather easy to fall into the trap of counting significant and nonsignificant G × E interactions and of letting the majority tip the balance. The narrative counting strategy has several shortcomings and risks. The most important one is that it heavily relies on null hypothesis significance testing (NHST), which during the past few years has once more come in for severe criticism from statistically sophisticated researchers (e.g., Cumming, Reference Cumming2014). Significance is largely a function of sample size, and NHST does not sufficiently take effect sizes and their confidence intervals into account. “Do not trust any p-value” is one of the Cumming (Reference Cumming2014) guidelines for new statistics.
Furthermore, several major journals now recommend less reliance on NHST in order to lower the risk of (unintentional) data manipulation triggered by dependence on the magic p < .05 (e.g., Psychological Science). For these reasons, the new statistical approaches proposed by Cumming (Reference Cumming2014) and others avoid all significance testing and rely on effect estimation and meta-analysis to document the replicability of any finding. Cumming (Reference Cumming2014) and many others emphasize using small- or large-scale meta-analysis to help avoid dependence on single studies, however impressive they might seem, and build a cumulative discipline based on replicated results. A quantitative or meta-analysis might generate insight in a replicable and responsible way because it allows for an estimate of the overall trend in the data and for an examination of study characteristics that may moderate the effect sizes. Thus, we decided to conduct a meta-analysis to examine whether the randomized G × E experiments reported in the current Special Section as well as in previous publications elsewhere support or refute differential susceptibility theory.
Types of G × E Experiments
Three types of G × E interventions might be differentiated, depending on the level of manipulation of the environment (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2015). First, nanotrials examine the immediate neural or behavioral responses to a small range of positive and negative stimuli, to minor manipulations of stress levels, or to subtle priming (e.g., Fox, Zougkou, Ridgewell, & Garner, Reference Fox, Zougkou, Ridgewell and Garner2011; Sasaki et al., Reference Sasaki, Kim, Mojaverian, Kelley, Young Park and Januŝonis2013). Nanotrials are meant to provide insight into mechanisms of change through a small window but with a sharp focus on part of the cascade of changes to be expected in broader trials.
Second, microtrials address a somewhat broader component of the environment but maintain a clear focus and modest manipulation of the environment. Consider in this regard evaluations of computerized early literacy instruction with and without personalized feedback (Kegel, Bus, & van IJzendoorn, Reference Kegel, Bus and van IJzendoorn2011; Plak, Kegel, & Bus, Reference Plak, Kegel and Bus2015 [this issue]) and the manipulation of social acceptance, rejection, stress, or retaliation to test its effects on aggression (Gallardo-Pujol, Andres-Pueyo, & Maydeu-Olivares, Reference Gallardo-Pujol, Andres-Pueyo and Maydeu-Olivares2013; McDermott, Tingley, Cowden, Frazzetto, & Johnson, Reference McDermott, Tingley, Cowden, Frazzetto and Johnson2009; Verona, Joiner, Johnson, & Bender, Reference Verona, Joiner, Johnson and Bender2006). Microtrials might be conducted to test proof-of-principle of whether a specific ingredient of broader and more ecological valid trials might contribute to its efficacy (see also Andersson et al., Reference Andersson, Ruck, Lavebratt, Hedman, Schalling and Lindefors2013; Soderqvist et al., Reference Söderqvist, Nutley, Peyrard-Janvid, Matsson, Humphreys and Kere2012).
Third, macrotrials, or field trials, are broad educational, parent training, or social programs that aim at changing general life circumstances for target groups including proximal and more distal components of their environment (Albert et al., Reference Albert, Dodge, Crowley, Bates, Pettit and Lansford2015 [this issue]; Bakermans-Kranenburg et al., Reference Bakermans-Kranenburg, van IJzendoorn, Pijlman, Mesman and Juffer2008; Beach, Brody, Lei, & Philibert, Reference Beach, Brody, Lei and Philibert2010; Bockting, Mocking, Lok, Koeter, & Schene, Reference Bockting, Mocking, Lok, Koeter and Schene2013; Brett et al., Reference Brett, Humphreys, Smyke, Gleason, Nelson and Zeanah2015 [this issue]; Brody, Chen, & Beach, Reference Brody, Chen and Beach2013; Brody, Chen, Beach, Kogan, et al. Reference Brody, Chen, Beach, Kogan, Yu and DiClemente2014; Brody et al., Reference Brody, Murry, Kogan, Gerrard, Gibbons and Molgard2006; Brody, Yu, & Beach, Reference Brody, Yu and Beach2015 [this issue]; Cicchetti, Rogosch, and Toth, Reference Cicchetti, Rogosh and Toth2011; Cleveland et al., Reference Cleveland, Schlomer, Vandenbergh, Feinberg, Greenberg and Spoth2015 [this issue]; Kohen et al., Reference Kohen, Cain, Buzaitis, Johnson, Becker and Teri2011; Van den Hoofdakker et al., Reference Van den Hoofdakker, Nauta, Dijck-Brouwer, Van der Veen-Mulders, Sytema and Emmelkamp2012). This is the type of trial that might be implemented on a large scale and is closest to the daily realities of parents, practitioners, or policymakers. In the current study, we examined whether genetic differential susceptibility can be found across the three types of trials, by contrasting the combined effect sizes of susceptible and nonsusceptible genotypes within macro-, micro-, and nanotrials.
Additional G × eE Moderators
Beyond type of trail, additional potential moderators of the effect sizes tested in the meta-analysis need to be considered. First, we tested whether experimental outcome was similar in G × E studies with mostly Caucasian versus mostly non-Caucasian participants. Ethnicity may play an important role in gene–environment interaction effects, as also evident from the divergent effects in studies with mostly (>80%) Caucasian participants versus studies with more mixed ethnicities involving the 5-HTT gene (van IJzendoorn et al., Reference van IJzendoorn, Belsky and Bakermans-Kranenburg2012; see also Propper, Willoughby, Halpern, Capone, & Cox, Reference Propper, Willoughby, Halpern, Capone and Cox2007; Williams et al., Reference Williams, Marchuk, Gadde, Barefoot, Grichnik and Helms2003; but see Vijayendran et al., Reference Vijayendran, Cutrona, Beach, Brody, Russell and Philibert2012).
Second, a number of intervention studies aimed at reducing aggressive and externalizing behavior, or at decreasing youth's alcohol or drug abuse, whereas other interventions aimed at decreasing internalizing symptomatology. Furthermore, some trials focused on promoting cognitive development. In the meta-analysis, we tested whether within these sets of intervention studies, the differential susceptibility effect (with larger combined effect sizes for the susceptible genotypes) can be found, to a larger or smaller extent.
Third, it is of interest to know whether specific candidate genes show larger differential susceptibility effects than others. It may be the case that some candidate genes make more of a difference for specific interventions or behavioral outcomes (e.g., monoamine oxidase A for aggression, 5-HTTLPR for depression); but there may also be a general trend for dopamine-related genes to show the largest differential susceptibility effect due to the engagement of the dopaminergic system in general attention, motivation, and reward mechanisms (Robbins & Everitt, Reference Robbins, Everitt and Zigmond1999).
Hypotheses
The first prediction is that the combination of the intervention effects across the G × eE RCTs for the carriers of the putative susceptible genotypes is significantly larger than the combined effect size shown for genotypes assumed to be associated with less susceptibility to environmental influences. Second, we expect that the more controlled experiments, that is, nanotrials, will show stronger differential susceptibility effects than the broader interventions, because the latter type of interventions might leave more room for error variance in the environmental component of the G × E equation. Third, we expect to find stronger intervention differences between more susceptible and less susceptible individuals in trials with predominantly Caucasian participants, because the usual genetic suspects of differential susceptibility mainly emerged from G × E studies on subjects of Caucasian ethnicity. Based on two previous meta-analyses on genetic differential susceptibility (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2011; van IJzendoorn et al., Reference van IJzendoorn, Belsky and Bakermans-Kranenburg2012), we predict stronger differential susceptibility for dopamine-system genes than for other genotypes. We do not have specific expectations for differences in effect sizes between studies aiming at decreasing externalizing behaviors versus those aiming at internalizing problems or cognitive delays.
Method
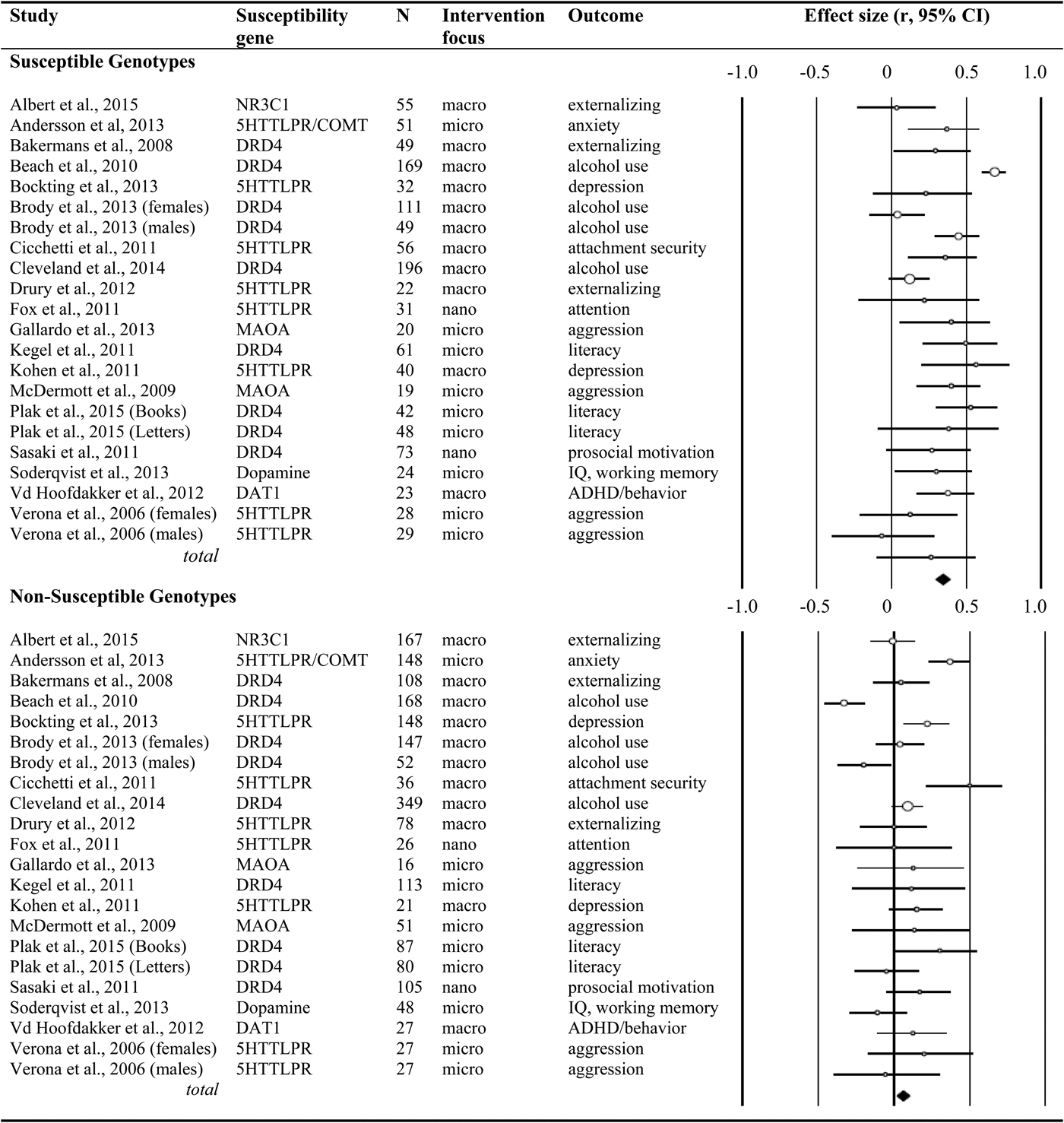
We systematically searched the databases Web of Science and MEDLINE, with the key words experiment*, genetic*, environment*, intervention*, random* trial, differential susceptibility, RCT, and G × E in the title or abstract (the asterisks indicate that the search contained the word or word fragment). The search was restricted to RCTs with humans, and we excluded pharmacological treatment (such as treatment with drugs, alcohol infusion, or oxytocin inhalation) as the experimental intervention. We finished the search in electronic databases in March 2014, but we also included the randomized trials reported in this Special Section of Development and Psychopathology on experimental genetic differential susceptibility in humans. The selected studies included polymorphisms in the serotonin and dopamine system genes, glucocorticoid receptor gene NR3C1, and monoamine oxidase A (see Figure 1).

Figure 1. Randomized controlled genetic differential susceptibility studies; characteristics and effect sizes for the susceptible and nonsusceptible genotypes.
We identified 22 experiments involving 3,257 participants. The Comprehensive Meta-Analysis program was used to transform the results of the individual studies into the common metric of correlations and to combine effect sizes (Borenstein, Hedges, Higgins, & Rothstein, Reference Borenstein, Hedges, Higgins and Rothstein2009; Borenstein, Rothstein, & Cohen, Reference Borenstein, Rothstein and Cohen2005). For every study, we computed the effect size of the experimental manipulation (i.e., experimental vs. control group) for subjects with the polymorphism considered indicative of heightened susceptibility (e.g., DRD4 seven-repeat allele, short variant of the 5-HTT gene) and for those with the genotype expected to convey low susceptibility (e.g., DRD4 four-repeat allele, long variant of the 5-HTT gene). In case of multiple outcome measures, effect sizes were combined within the study before adding the resulting effect size to the final meta-analyses. In case of multiple time points with the same outcome assessments, we selected the time point closest to the end of the intervention, to enhance comparability of effect sizes across studies.
Heterogeneity across sets of outcomes was assessed using the Q-statistic. Because most of our data sets were heterogeneous in their effect sizes and because random effects models are somewhat more conservative than fixed effects parameters in such cases, combined effect sizes and confidence intervals from random effects models are presented. We tested the influence of genotypes as susceptibility markers on the variation in combined effect sizes with the Q contrast statistic in a random effects model. The Q contrast statistic is based on the logic of analysis of variance, with the total variance Q total partitioned into Q between and Q within; Q total is the variance with any grouping factors ignored; and Q within for each group refers to the variances in the specific subsets of outcomes. The Q within for the susceptible group thus reflects the variance in intervention effects across all studies for the subgroup with the assumed susceptible genotypes, and the Q within for the nonsusceptible group reflects the variance in intervention effects across all studies for the subgroup with the nonsusceptible genotypes. Here, Q between = Q total – Q within and is tested for significance using the chi-square distribution (Borenstein et al., Reference Borenstein, Hedges, Higgins and Rothstein2009). A significant Q contrast value indicates that the difference in effect size between susceptible genotypes and nonsusceptible genotypes is significant. We tested the significance of Q contrast for the total set of RCTs and for specific subgroups of studies.
Following the overall contrast, we tested whether the contrast was similar in studies with mostly Caucasian participants. Next, we examined whether genetic differential susceptibility could be found across the three types of trials, by contrasting the combined effect sizes of susceptible and nonsusceptible genotypes within macro-, micro-, and nanotrials. A number of intervention studies sought to reduce or prevent the development of aggressive and externalizing behavior, or hoped to decrease youth's alcohol consumption. Another set of studies aimed at decreasing internalizing symptoms. Yet another set of studies focused at improving cognitive development. We tested whether within these sets of intervention studies the differential susceptibility effect (with larger combined effect sizes for the susceptible genotypes) would emerge. Finally, we explored whether specific candidate genes (i.e., dopamine-related genotypes) showed larger differential susceptibility effects than others (e.g., serotonin-related genotypes). In some cases, more than one genotype was tested for differential susceptibility, in which case the primary marker was selected (e.g., DRD4; Cleveland et al., Reference Cleveland, Schlomer, Vandenbergh, Feinberg, Greenberg and Spoth2015 [this issue]). Moderators were coded by two coders (intercoder reliability κ > 0.80); discrepancies were discussed, and consensus codes were used.
Finally, we computed the effect sizes for the difference in intervention effect between the susceptible and nonsusceptible genotypes within each of the RCTs. Positive effect sizes point to a larger effect for the susceptible genotypes than for the nonsusceptible genotypes. The overall combined effect size then reflects the susceptibility effect across studies.
In addition, we tested whether the distribution of these effect sizes showed any publication bias (Borenstein, Reference Borenstein, Rothstein, Sutton and Borenstein2005) favoring the publication of studies with larger differential susceptibility effects in smaller samples with the “trim and fill” method (Duval & Tweedie, Reference Duval and Tweedie2000a, Reference Duval and Tweedie2000b). Using this method, a funnel plot is constructed of each study's effect size against the sample size or the standard error (usually plotted as 1/SE, or precision). It is expected that this plot has the shape of a funnel, because studies with smaller sample sizes and larger standard errors have increasingly large variation in estimates of their effect size as random variation becomes increasingly influential, whereas studies with larger sample sizes have smaller variation in effect sizes (Duval & Tweedie, Reference Duval and Tweedie2000b; Sutton, Duval, Tweedie, Abrams, & Jones, Reference Sutton, Duval, Tweedie, Abrams and Jones2000). The plots should be shaped like a funnel if no data censoring is present. We used Egger's test for detecting funnel plot asymmetry. Because smaller nonsignificant studies are less likely to be published (the “file-drawer” problem; Mullen, Reference Mullen1989), studies in the bottom left-hand corner of the plot are often omitted (Sutton et al., Reference Sutton, Duval, Tweedie, Abrams and Jones2000). The studies considered to be symmetrically unmatched can then be trimmed; that is, their missing counterparts can be imputed or “filled” as mirror images of the trimmed outcomes, allowing for the computation of an adjusted overall effect size and confidence interval (Gilbody, Song, Eastwood, & Sutton, Reference Gilbody, Song, Eastwood and Sutton2000; Sutton et al., Reference Sutton, Duval, Tweedie, Abrams and Jones2000).
Results
Overall effect sizes
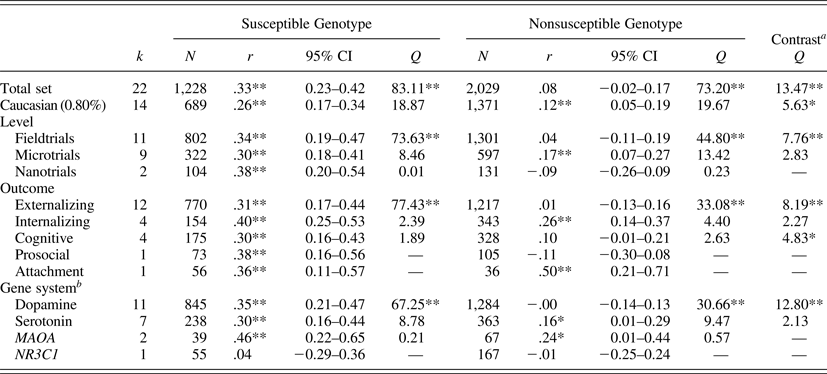
We identified 22 studies, including N = 3,257 participants, 1,228 of whom were carriers of vulnerability or rather susceptibility genes. The combined effect size of the intervention effects in this susceptible group yielded a Pearson r = .33 (95% confidence interval [CI] = 0.23, 0.42; p < .01); see Table 1. The nonsusceptible group consisted of 2,029 cases, and the combined size of the intervention effects in this group was not significant, r = .08 (95% CI = −0.02, 0.17; p = .12). Formal test of the contrast between the two combined effect sizes yielded a Q (1) = 13.47, p < .01, showing much stronger effects for the susceptible group.
Table 1. Combined effect sizes for the susceptible and nonsusceptible genotypes in Gene × Environment experiments
Note: k, Number of study outcomes; N, total sample size; d, effect size (Cohen d); 95% CI, 95% confidence interval around the point estimate of the effect size; Q homogeneity, homogeneity statistic; Q contrast, moderation statistic.
aSubgroups with k < 4 excluded from contrast.
bAndersson et al. (Reference Andersson, Ruck, Lavebratt, Hedman, Schalling and Lindefors2013) reported on combined dopamine/serotonin system genes.
*p < .05. **p < .01. ***p < .001.
Ethnicity
In the 14 studies with more than 80% Caucasian participants (n = 689 susceptible, n = 1,371 nonsusceptible), we found basically the same results, with significantly larger intervention effects for the susceptible genotypes, r = .26 (95% CI = 0.17, 0.34; p < .01) than for the nonsusceptible genotypes, r = .12 (95% CI = 0.05, 0.19; p < .01). These two sets of studies were both homogeneous, and the contrast was significant, Q (1) = 5.63, p = .02. The 8 studies with less than 80% Caucasian participants were too heterogeneous in terms of ethnicity to be combined in separate analyses.
Intervention focus
Eleven studies were macrotrials; they were significantly more effective in carriers of susceptible genotypes (r = .34, 95% CI = 0.19, 0.47; p < .01) than in carriers of nonsusceptible genotypes (r = .04, 95% CI = −0.11, 0.19; p = .60); contrast Q (1) = 7.76, p < .01 (see Figure 2). The difference among the nine microtrials was not significant (susceptible groups r = .30, 95% CI = 0.18, 0.41; p < .01, nonsusceptible groups r = .17, 95% CI = 0.07, 0.27; p < .01); contrast Q (1) = 2.83, p = .09. The two nanotrials showed larger effects in susceptible genotype groups (r = .38, 95% CI = 0.20, 0.54, p < .01) than in nonsusceptible genotypes (r = −.09, 95% CI = −0.26, 0.09, p = .32), though the contrast between the two groups could not be tested due to the small number of studies.
Figure 2. (Color online) Combined effect sizes (r) for the susceptible and nonsusceptible genotypes in macrotrials, microtrials, and nanotrials.
Behavioral outcome
Twelve interventions targeted externalizing behaviors (including alcohol abuse). Carriers of susceptible genotypes were significantly more affected, Q (1) = 8.19, p < .01, by the interventions (r = .31, 95% CI = 0.17, 0.44, p < .01) than their nonsusceptible peers (r = .01, 95% CI = −0.13, 0.16, p = .87). The four interventions targeting internalizing behaviors were effective both in the susceptible genotypes (r = .40, 95% CI = 0.25, 0.53, p < .01) and in the nonsusceptible genotypes (r = .26, 95% CI = 0.14, 0.37, p < .01), and the contrast was not significant, Q (1) = 2.27, p = .13. Interventions in the cognitive domain showed larger effects in susceptible genotypes (r = .30, 95% CI = 0.16, 0.43, p < .01) than in nonsusceptible genotypes (r = .10, 95% CI = −0.01, .21, p = .06), and the contrast was significant, Q (1) = 4.83, p < .05.
Susceptibility genes
Considering the genetic marker of susceptibility, dopamine-related genes were markers of susceptibility; the eleven studies with dopamine-related genotypes as moderator showed larger intervention effects in susceptible genotype groups (r = .35, 95% CI = 0.21, 0.47, p < .01) than in nonsusceptible genotypes (r = −.00, 95% CI = −0.14, 0.13, p = .96); the contrast was significant, Q (1) = 12.80, p < .01. Seven studies with 5-HTTLPR as moderator showed significant combined effects in the susceptible genotype group (r = .30, 95% CI = 0.16, 0.44, p < .01) but also in the nonsusceptible genotype (r = .16, 95% CI = 0.01, 0.29, p = .04); the contrast was not significant, Q (1) = 2.13, p = .15.
Within-study differential susceptibility effects
As a final step, we computed the difference between the Fisher Z-transformed effect sizes for the susceptible and nonsusceptible groups within each study. The combined effect size was Fisher Z = 0.23 (95% CI = 0.09, 0.37; p < .01), showing a significant combined effect for the difference between susceptible and nonsusceptible genotypes. The funnel plot of these effect sizes did not show publication bias; thus trim-and-fill was not necessary, and the Egger's test was not significant (t = 0.54, p = .30).
Discussion
Clear-cut experimental support emerged for genetic differential susceptibility. In our meta-analysis on 22 RCTs including 3,257 participants, 38% of whom were carriers of susceptibility genes, the combined effect size of the interventions for the carriers of the susceptible genotypes amounted to r = .33. This is a large effect even in terms of Cohen's (Reference Cohen1988) conventional criteria. In contrast, the hypothesized nonsusceptible group did not appear to be affected by the interventions. The two earlier meta-analyses on genetic differential susceptibility, including mostly correlational studies instead of RCTs yielded remarkably convergent evidence (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2011; van IJzendoorn et al., Reference van IJzendoorn, Belsky and Bakermans-Kranenburg2012), thus demonstrating the generativity and emerging validity of genetic differential susceptibility theory.
From the perspective of science or intervention practice, it might be too early to consider the available evidence for genetic differential susceptibility sufficient. Replication is paramount in science (Cumming, Reference Cumming2014), and although the number of replicating studies is rapidly increasing, there is still some way to go, perhaps most important in terms of further understanding the biological and psychological mechanisms of differential change. Sound intervention practice also requires the most firm scientific foundation, and the number of populations and environments covered in the current set of studies is limited to small part of the Western, educated, industrialized, rich, and democratic societies (Henrich, Heine, & Norenzayan, Reference Henrich, Heine and Norenzayan2010).
Nevertheless, the finding of a robust genetic differential susceptibility effect is of great importance for at least two reasons. First, experimental G × E studies exclude or control for several alternative interpretations that have plagued correlational G × E studies, such as undetected rGE or lack of power and thus replicability. Second, they make clear that even in the absence of overall efficacy, interventions should not be discarded as ineffective and useless because they may have large impact on the substantial minority of more susceptible participants. Differential susceptibility theory allows for theory-guided examination of moderators because a small set of potential differential susceptibility markers have been proposed: biological sensitivity to context, temperamental reactivity, and genetic markers, in particular related to the dopaminergic and serotonergic systems (Ellis, Boyce, Belsky, Bakermans-Kranenburg, & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2011).
Ethnicity
Population stratification is a potential threat to the validity of genetic findings, which is why we repeated the meta-analysis in the largest and ethnically most homogeneous set of 14 studies with more than 80% Caucasian participants. Genetic differential susceptibility was clearly present in this set, but it was not possible to examine differential susceptibility in other ethnicities due to the small numbers of studies. Because the difference in effect sizes between the Caucasian carriers of susceptibility genotypes and the non-Caucasian carriers of susceptibility genotypes was small, we presume that the genetic differential susceptibility effect is not restricted to individuals with Caucasian ethnicity (see, e.g., Brody et al., Reference Brody, Yu and Beach2015 [this issue], on African American families). Nevertheless, it is important to keep in mind that the biological functionality of genotypes have been shown to be different across ethnicities, and therefore, similar alleles might be responsible for contrasting levels of neurotransmitters, enzymes, or hormones regulating information processing and behavior. Besides using methods such as principal coordinates analysis to assess and control for population stratification (Cleveland et al., Reference Cleveland, Schlomer, Vandenbergh, Feinberg, Greenberg and Spoth2015 [this issue]), testing for the robustness of differential susceptibility evidence in ethnically homogeneous subgroups of the study remains recommended.
Focus and outcome
It should be noted that genetic differential susceptibility was documented more firmly in macrotrials than in microtrials and nanotrials, despite the hypothesized large amount of error variance in the treatment manipulation in macrotrials. Moreover, it was more difficult to demonstrate differential susceptibility for internalizing outcomes than for externalizing and cognitive outcomes. Of course, this pattern of results is partly due to the smaller number of studies, as in the cases of nanotrials and the focus on depression and anxiety outcome measures. With the current small set of studies, it is impossible to tease apart the moderating effects (or absence thereof) of focus and outcome because they might be intertwined, with macrotrials more often aiming at externalizing issues, and micro- and nanotrials more often focusing on cognition or anxiety. However, we should be aware of the possibility that with some types of interventions (microtrials) or specific outcomes (internalizing symptoms) the impact on carriers of the less susceptible genotypes might be too large to be exceeded by the impact on the susceptible participants. In a similar vein, some interventions might be so effective that every subject profits, especially when these subjects have been living in extremely bad environments (e.g., maltreating families; Cicchetti et al., Reference Cicchetti, Rogosh and Toth2011). The G × E equation underlying genetic differential susceptibility illustrates that an extremely large (change of the) E component might drown out the influence of the G component.
Dopamine or serotonin?
The majority of G × E experiments targeted dopamine-related genes as the genetic markers of susceptibility, and they appeared to moderate intervention efficacy. This was not the case in the smaller set of studies focusing at the serotonin transporter gene (5-HTTLPR) as moderator. We found a similar trend in the two previous meta-analyses on mostly correlational G × E studies. In the meta-analysis on dopamine-related genotypes, differential susceptibility was more pronounced (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2011) than in the meta-analysis on serotonin-related genotypes (van IJzendoorn et al., Reference van IJzendoorn, Belsky and Bakermans-Kranenburg2012), although in the latter case the set of Caucasian samples showed significant differential susceptibility. In the current meta-analysis on G × E experiments, carriers of the putatively more susceptible 5-HTT short alleles profited substantially from the interventions, but their counterparts with the long alleles also profited, albeit to a lesser extent. The difference was too small to be significant.
It is too early to draw conclusions from this trend because it is based on only 11 and 7 RCTs, but if confirmed in subsequent investigations, one of the alternative interpretations might reside in epigenetic change. In a study on traumatic childhood experiences and adult functioning, we found that increased methylation of the long 5-HTT alleles made their carriers more vulnerable to symptoms of unresolved loss and posttraumatic stress (van IJzendoorn, Caspers, Bakermans-Kranenburg, Beach, & Philibert, Reference van IJzendoorn, Caspers, Bakermans-Kranenburg, Beach and Philibert2010), and thus more similar to carriers of the short variants. Different genotypes might be differentially open to (de)methylation, and epigenetic changes influencing the expression of genetic markers of differential susceptibility might have to be taken into account when conducting G × E trials (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2015; Meaney, Reference Meaney2010).
Statistics
Various statistical approaches have been developed to examine the precise shape of G × E interactions and to decide whether interactions fit diathesis–stress or differential susceptibility models. One of them uses the regions of significance (Kochanska, Kim, Barry, & Philibert, Reference Kochanska, Kim, Barry and Philibert2011), another the proportion of interaction or the proportion affected (Roisman et al., Reference Roisman, Newman, Fraley, Haltigan, Groh and Haydon2012), and yet another method was developed by Widaman et al. (Reference Widaman, Helm, Castro-Schilo, Pluess, Stallings and Belsky2012). In this approach, the predictor is centered at the crossover point, and a confidence interval for the crossover point is estimated. When both the crossover point and its confidence interval fall within the range of observed predictor values, the interaction represents differential susceptibility; when both fall outside the range of observed predictor values, the interaction suggests diathesis stress. The strong version of differential susceptibility implies that those who are not susceptible are not at all affected by the environmental predictor; the weak version implies that some are less affected than others. The advantage of this approach is the formal testing of nested diathesis–stress and differential susceptibility models (Belsky, Pluess, & Widaman, Reference Belsky, Pluess and Widaman2013).
Although developed for correlational G × E designs, we propose that it can be used with G × E experiments as well. Using this method, we tested the results of our trial with video-feedback parenting training resulting in lower levels of daily cortisol production in toddlers with the DRD4 seven-repeat allele (Bakermans-Kranenburg et al., Reference Bakermans-Kranenburg, van IJzendoorn, Pijlman, Mesman and Juffer2008). The estimated crossover point and its confidence interval fell completely within the range of the dichotomous predictor (intervention or control group), convergent with differential susceptibility. Moreover, relaxing the constraint that the effect of the intervention for the nonsusceptible group was zero did not improve model fit significantly, and it could thus be concluded that the data supported the strong version of the differential susceptibility model (see Plak et al., Reference Plak, Kegel and Bus2015 [this issue]). This example demonstrates that G × E trials may profit from the application of the Widaman et al. (Reference Widaman, Helm, Castro-Schilo, Pluess, Stallings and Belsky2012) model-fitting approach.
Misunderstandings
Some critical misunderstandings about G × E trials need to be discussed. The first misunderstanding is the assumption that to find a replicable G × E interaction, one would need a genetic main effect (e.g., Munafò Zammit, & Flint, in press; Risch et al., Reference Risch, Herrell, Lehner, Liang, Eaves and Hoh2009). Rutter, Thapar, and Pickles (Reference Rutter, Thapar and Pickles2009) listed three reasons for doubting the soundness of this assumption. First, a crossover interaction is not accompanied by a main effect if the crossover point is in the middle of the environmental continuum. Second, if G × E is found in individuals without psychopathology, a main genetic effect on psychopathology might be absent. Third, statisticians have not finished debating the pros and cons of the assumption from a purely statistical point of view, and no consensus has been reached. Elsewhere we argued that the assumption is a logical implication of a diathesis–stress or vulnerability viewpoint, which becomes obsolete in a differential susceptibility perspective (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2015). Differential susceptibility theory implies that environmental effects are small or absent for one genotype, but present for the other genotype, for better and for worse. In that case, the two directions within one genotype cancel each other out and significant G × E effects are found in the absence of a genetic main effect. Similarly, the overall efficacy of interventions (main effect) might go undetected because it is hidden in a G × E interaction in a “for better and for worse” fashion.
A second misunderstanding is involved in the application of the so-called vantage sensitivity concept (Manuck & McCaffery, Reference Manuck and McCaffery2014; Pluess & Belsky, Reference Pluess and Belsky2013; Sweitzer et al., Reference Sweitzer, Halder, Flory, Craig, Gianaros and Ferrell2012) to experimental G × E studies. To characterize the “bright side” of differential susceptibility (Bakermans-Kranenburg & van IJzendoorn, Reference Bakermans-Kranenburg and van IJzendoorn2011), the term vantage sensitivity (Manuck & McCaffery, Reference Manuck and McCaffery2014; Sweitzer et al., Reference Sweitzer, Halder, Flory, Craig, Gianaros and Ferrell2012) was introduced to suggest that some individuals profit more than others from supportive environments but adapt to negative environments in similar ways as their genotypic counterparts (Pluess & Belsky, Reference Pluess and Belsky2013). When in a randomized trial at baseline carriers of susceptible and nonsusceptible genotypes show the same number of externalizing, internalizing, or cognitive problems, as one would hope in a sound RCT (for examples, see Kegel et al., Reference Kegel, Bus and van IJzendoorn2011; Plak et al., Reference Plak, Kegel and Bus2015 [this issue]), a positive outcome of the intervention might be automatically interpreted as an example of vantage sensitivity. However, this is a non sequitur because in genetic differential susceptibility experiments the genetic moderators have been chosen because they were markers of differential susceptibility in correlational G × E studies, and considered risk genes in the first place. Furthermore, the vantage interpretation of positive outcomes in a positive environment in a G × E trial would suffer from the same problem as interpreting negative outcomes in a negative environment in terms of diathesis stress in a correlational study: both may be based on a truncated continuum of environments and through a narrow window are only able to see one side of the equation, dark or bright, but not both.
Limitations
An obvious limitation of the current meta-analysis is the relatively small number of studies. This may reflect that RCTs require almost heroic investments in time and other resources, and that the rewards may be somewhat disappointing, for example, in terms of impact and number of publications. Nevertheless, more genetic RCTs are badly needed. The prevailing candidate gene approach in G × E experiments may be complemented by approaches that use biologically functional genetic pathways instead of single genes to serve as markers of differential susceptibility.
Moreover, we need more experimental work with within-subject designs. A critical assumption of differential susceptibility is that the same individual would be more or less open to environmental pressures, for better and for worse. No G × E experiments have used designs in which the same individual is exposed both to supportive and to negative conditions, for obvious ethical reasons. However, micro- or nanotrials using mildly negative and positive manipulations, as in attention bias modification trials, may provide crucial evidence for this core differential susceptibility hypothesis (e.g., Fox et al., Reference Fox, Zougkou, Ridgewell and Garner2011; Hakamata et al., Reference Hakamata, Lissek, Bar-Haim, Britton, Fox and Leibenluft2010). Furthermore, more sophisticated treatment of covariates may be necessary (Keller, Reference Keller2014; for an example, see Cicchetti, Toth, & Handley, Reference Cicchetti, Toth and Handley2015 [this issue]).
A final limitation is the sparsely available evidence on biological and psychological mechanisms responsible for differential susceptibility. We need more insight into mediators that transfer intervention influences to outcomes. Genetically moderated intervention efficacy is always moderated mediation, and the search for pertinent mediators should be prioritized (for an example, see Brody et al., Reference Brody, Yu and Beach2015 [this issue]).
Implications
If the evidence for genetic differential susceptibility proves to be solid, it will have far-reaching consequences (see Bakermans-Kranenburg & van IJzendoorn, in press; for a more detailed discussion, see also Ellis et al., Reference Ellis, Boyce, Belsky, Bakermans-Kranenburg and van IJzendoorn2011). It may not only radically change our view of risk genotypes and related bad phenotypes (Belsky et al., Reference Belsky, Bakermans-Kranenburg and van IJzendoorn2007; Belsky & Pluess, Reference Belsky and Pluess2009, Reference Belsky and Pluess2013; Ellis et al., Reference Ellis, Boyce, Belsky, Bakermans-Kranenburg and van IJzendoorn2011) but also contribute to the solution of a tenacious problem, that is, the missing heritability in worldwide immensely large genome-wide association studies failing to find substantial genetic main effects on complex traits and disorders (Plomin, Reference Plomin2013). Even genome-wide complex trait analysis (Yang, Lee, Goddard, & Visscher, Reference Yang, Lee, Goddard and Visscher2011), a promising new tool to estimate heritability, still leaves a considerable gap with behavioral genetics estimates of the same phenotypes (Trzaskowski, Dale, & Plomin, Reference Trzaskowski, Dale and Plomin2013). Missing heritability might be caused by the neglect of environmental influences, or better: G × E effects that are absorbed in the additive genetic part of the pie in many twin studies and completely neglected in genome-wide association studies or genome-wide complex trait analysis studies (van IJzendoorn et al., Reference van IJzendoorn, Bakermans-Kranenburg, Belsky, Beach, Brody and Dodge2011). Experimental manipulation of E might be the best way to show that complex human behavior can only be understood as the outcome of G × E interplay, in support of the famous dictum by Bronfenbrenner (Reference Bronfenbrenner1979) that main effects will be shown to reside in interactions.
We conclude that the available G × E experiments on more than 3,000 subjects provide replicable support for genetic differential susceptibility across a number of intervention modalities and target populations. This is a rich harvest after less than a decade since the first G × E experiment in the domain of human development demonstrating genetic differential susceptibility (Bakermans-Kranenburg et al., Reference Bakermans-Kranenburg, van IJzendoorn, Pijlman, Mesman and Juffer2008). The combined RCTs have provided proof of principle that genetic differential susceptibility exists, and it is now time to explore its mechanisms and limits. The concept of differential susceptibility has already altered the way in which we interpret so-called weakly effective or ineffective (preventive) interventions, because it teaches us to look beyond intervention main effects into the hidden interactions.