



We read with great interest the recent Canadian Guidelines for Hereditary Transthyretin Amyloidosis Polyneuropathy Management, Reference Alcantara, Mezei and Baker1 which provides evidence-based recommendations in the diagnosis, treatment, and surveillance of hereditary transthyretin amyloidosis (hATTR) polyneuropathy (PN). As mentioned in the guidelines, patients with late-onset hATTR PN present with length-dependent sensory disturbance involving pansensory loss, typically associated with pain, and which progresses to loss of deep tendon reflexes, distal weakness, imbalance, and autonomic dysfunction. Reference Alcantara, Mezei and Baker1,Reference Dohrn, Rocken and De Bleecker2
One diagnostic challenge with late-onset hATTR PN is differentiating the nature of the neuropathy in the presence of competing comorbidities. Up to one-third of individuals over the age of 65 have a diagnosis of diabetes, and a large proportion are undiagnosed. Reference Corriere, Rooparinesingh and Kalyani3 Moreover, in a population-based study aimed at determining the prevalence of monoclonal gammopathy of undetermined significance (MGUS), it was found that MGUS increases with age, with a prevalence of 4.7% between the fifth and sixth decades of life, increasing to 11.2% in individuals over the age of 70. Reference Kyle, Therneau and Rajkumar4 Further, in a case series of 15 patients with hATTR PN, coincident diabetes mellitus and monoclonal gammopathy occurred in 23% and 7%, respectively. Reference Dohrn, Rocken and De Bleecker2 Therefore, the traditional nerve biopsy can provide diagnostic clarity in the setting of multiple competing factors, but on the other hand, the guidelines prudently highlight the drawbacks of the nerve biopsy, which include potential risk of infection, sample limitation, and a high proportion of false negatives. Reference Alcantara, Mezei and Baker1
We are reminded of the value of nerve biopsy by a patient we encountered just weeks prior to the publication of the guidelines – a 74-year-old male of Jamaican ethnicity and a history of poorly controlled insulin-dependent diabetes, dyslipidemia, hypertension, and bilateral carpal tunnel syndrome. He was evaluated by cardiology for progressive fatigue and lightheadedness, which revealed NYHA Class II heart failure due to restrictive cardiomyopathy and an intermittent high-degree AV block requiring implantation of a pacemaker. Further investigations uncovered an IgG kappa light-chain monoclonal gammopathy with a negative bone marrow biopsy for plasma cell dyscrasia. Mass spectroscopic analysis of a right ventricle biopsy demonstrated transthyretin amyloidosis, for which subsequent genetic testing confirmed to be the Val142Ile variant.
The patient had experienced 5 years of distal lower extremity numbness, which was felt to be diabetic neuropathy. Electrodiagnostic examination confirmed an axonal sensorimotor polyneuropathy. However, given the co-existing diabetes, MGUS, and hATTR, a left sural nerve biopsy was performed to help clarify the etiology of his polyneuropathy.
Pathological examination of the nerve revealed a primary axonal neuropathy with possible secondary demyelination, axonal regeneration, and remyelination (Figure 1A–J). Amyloid deposition was identified in the endoneurium as lobulated areas consisting of amyloid fibrils on semi-thin sections with electron microscopy (EM) (Figure 1E–H). Amyloid deposition was not in the extraneurial connective tissue, blood vessels, or the portion of formalin-fixed paraffin-embedded tissue, as demonstrated with negative Congo red staining (Figure 1D). The endoneurial capillaries were markedly thickened with multiple concentric layers of basal lamina (Figure 1C, K, L), with some thickened perineurial blood vessels and perineurial basal laminae. Immunohistochemistry showed mild perivascular infiltration of CD3+ T-cells, absence of CD20+ B-cells, and CD138+ plasma cells, with nonspecific positivity of kappa and lambda immunostaining (without evidence of monoclonality or light-chain amyloidosis), and negative β-amyloid. Taken together, the mixed axonal degeneration-regeneration and demyelination-remyelination, with endoneurial amyloid deposition and associated vascular changes, are consistent with co-existing hATTR amyloidosis and diabetic polyneuropathy.
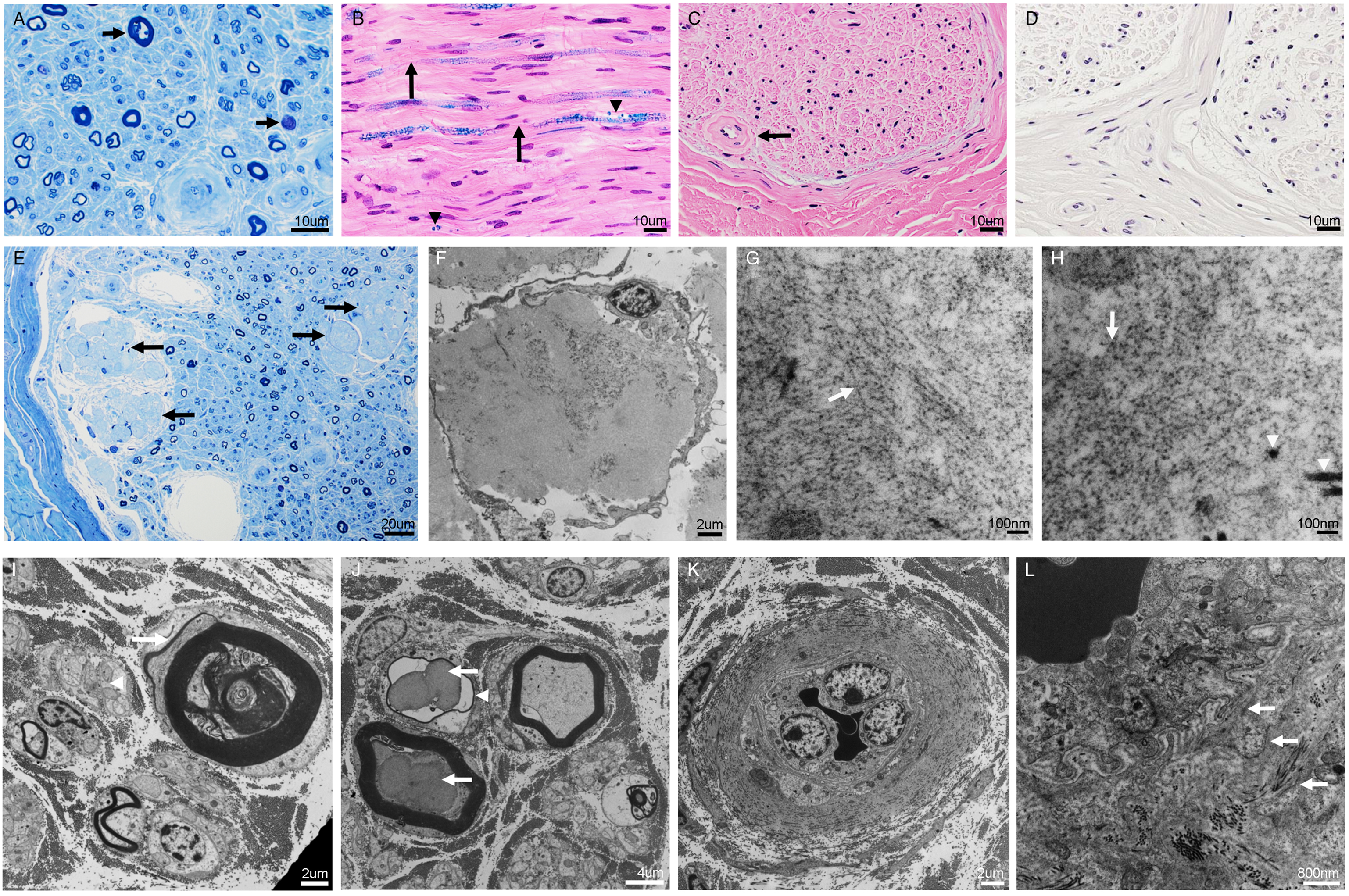
Figure 1: Microphotographs demonstrating comorbid amyloid and diabetic neuropathy in a sural nerve biopsy. The nerve shows moderate loss of myelinated and unmyelinated fibers, axonal degeneration with myelin ovoids (A, arrows; semi-thin section, toluidine blue staining), possible secondary demyelination (B, arrows; Luxol fast blue and Hematoxylin–Eosin staining) with associated axonal degeneration and myelin ovoids (B, arrowheads), concentric thickening of endoneurial capillaries (C, arrow; Hematoxylin–Eosin staining), with negative Congo Red staining (D), and endoneurial lobulated deposits of amorphous material (E, arrows; semi-thin section, toluidine blue staining). Electron microscopy revealed that the endoneurial lobulated deposits of amorphous material (F) are composed of irregular, unbranched amyloid fibrils on a longitudinal section (G, arrow pointing at amyloid fibrils) and on a cross section (H, arrow pointing at amyloid fibrils compared to arrowheads pointing at larger collagen fibrils), measuring 9–12 nm in diameter. There are also associated atrophic and degenerative axons with myelin ovoid and peeling myelin suggestive of secondary demyelination (I, arrow pointing at a degenerative axon with peeling myelin), in addition to axonal sprouting/regeneration (I, arrowhead), degenerative lobulated axons (J, arrows), thinly myelinated fiber suggestive of remyelination (J, arrowhead), and capillary thickening (K) with multiple concentric layers of basal lamina (L, arrows) but no amyloid deposition (L and D). Scale bars: 10 µm (A–D), 20 µm (E), 2 µm (F, I, K), 100 nm (G, H), 4 µm (J), 800 nm (L).
Given that the patient’s cardiac biopsy demonstrated transthyretin amyloidosis, he qualified for the use of the transthyretin tetramer stabilizer, Tafamidis. However, he was not yet initiated on Tafamidis. In light of his nerve pathology being consistent with hATTR amyloidosis, the treatment options for his hATTR amyloidosis expanded to include the potential use of patisiran, a small-interfering RNA that reduces the production of transthyretin that has been shown to have both favorable neuropathy and cardiac benefits. Reference Alcantara, Mezei and Baker1
Hereditary ATTR is an under-recognized multisystemic disease with heterogenous clinical manifestations that can lead to significant morbidity and early mortality if untreated. Reference Alcantara, Mezei and Baker1,Reference Galant, Westermark, Higaki and Chakrabartty5,Reference Plante-Bordeneuve and Said6 Certain pathogenic mutations in the transthyretin gene have been identified to be predominantly associated with a cardiac versus neuropathic phenotype. Reference Alcantara, Mezei and Baker1,Reference Maurer, Hanna and Grogan7 The Val142Ile mutation is one of the most common forms of cardiac amyloidosis and often presents in individuals of African ethnicity. Less commonly, patients also experience neuropathic symptoms and walking disability. Reference Maurer, Hanna and Grogan7 Despite this, little is known about the associated nerve pathology, although our findings appear to be consistent with prior studies which also demonstrated endoneurial amyloid deposition. Reference Dohrn, Rocken and De Bleecker2,Reference Devarapalli, Zhou, Dyck and Piccione8 Interestingly, previous studies revealed that the majority (˜70%) of nerve biopsy specimens from genetically confirmed hATTR patients demonstrated amyloid deposition, but the remaining 17%–30% had no amyloid deposition detected by light microscopy histological examination with negative Congo red staining. Reference Dohrn, Rocken and De Bleecker2,Reference Luigetti, Romozzi and Bisogni9 Therefore, the inability to find amyloid deposition with Congo red staining in a nerve biopsy specimen does not rule out the diagnosis of hATTR PN. Instead, further EM examination may demonstrate unbranched amyloid fibrils and minute deposits. Reference Dohrn, Rocken and De Bleecker2 In our study, despite the negative Congo red staining within the tissue portion used for light microscopy histological examination, another tissue portion subject to semi-thin section for EM analysis was able to exhibit endoneurial lobulated deposits of amorphous amyloid material consistent with amyloid fibrils, which prior studies have also demonstrated. Reference Dohrn, Rocken and De Bleecker2,Reference Devarapalli, Zhou, Dyck and Piccione8 It is important to keep in mind that amyloid fibrils in nerve biopsy specimens analyzed on EM are morphologically heterogenous and differ, not only based on the underlying transthyretin gene variant, but also the patient’s age, whether the patient is from an endemic or non-endemic area, and phase of amyloid fibril formation. Reference Koike, Nishi and Ikeda10 As such, given the findings from the EM analysis, the patient’s negative bone marrow biopsy for plasma cell dyscrasia and confirmed genetic testing for the Val142Ile variant for hATTR, we felt that a diagnosis of hATTR PN could be made.
Given that hATTR is a multisystemic disease that frequently exists with competing diseases that can cause overlapping peripheral nerve pathologies, determining the nature of nerve involvement is important, as it can impact therapeutic decisions and prognosis. Therefore, our case demonstrates the value of the traditional nerve biopsy in the work-up of genetically confirmed hATTR.
Acknowledgements
The authors would like to thank Ms. Marilyn Timleck for performing the electron microscopy.
Statement of Authorship
GM and J-QL contributed to writing and reviewing the manuscript. MC contributed to the review of the manuscript. All authors were involved in the patient’s circle of care.
Conflict of Interest
All authors declare that they have no conflicts of interest.

