


Introduction
Interleukin-6 (IL-6), a member of the pleiotropic cytokine family, has complex effects on the central nervous system (CNS) (Reference Gadient and Otten1). Under normal conditions, the production of IL-6 in the CNS is highly regulated (Reference Hopkins and Rothwell2,Reference Van Wagoner and Benveniste3). In neural functional disorders, such as spinal cord diseases and injuries, IL-6 expression increases remarkably (Reference Kaplin, Deshpande and Scott4,Reference Brisby, Olmarker and Larsson5). The increased IL-6 may reflect a harmful process as an injurious mediator and a negative effect on neuroprotection. For example, IL-6 is a detrimental player contributing to the pathogenesis of many spinal cord diseases, for example, cervical spondylotic myelopathy and spinal cord injury (Reference Kanemoto6–Reference Neuhuber, Timothy Himes, Shumsky, Gallo and Fischer8). However, the increase of IL-6 may also represent a compensative mechanism for neural repair. For instance, IL-6 regulates neuronal function and development in the innate response of the CNS to injuries and diseases (Reference Sallmann, Juttler and Prinz9,Reference Jones, McDaniel and Popovich10), and exerts neurotropic and neuroprotective effects on glutamate- and N-methyl-d-aspartic acid (NMDA)-induced neuronal damage (Reference Carlson, Wieqqel, Chen, Bacchi, Roqers and Gahring11–Reference Wang, Peng and Lu14). These controversial functions of IL-6 in the CNS suggest the need for an extensive investigation of its role in spinal cord physiology and pathology.
Voltage-gated sodium channels, which are dynamic transmembrane proteins consisting of a pore-forming α-subunit (220–260 kDa) and auxiliary β-subunits (32–36 kDa), mediate the inward sodium currents of excitable cells and are thus key regulators of action potential (AP) generation and propagation (Reference Hodgkin and Huxley15–Reference Zhou, Qi and Zhao17). It has been reported that in the fibroblast growth factor homologous factors knockout mice, the electrophysiology properties of voltage-gated sodium channels are changed, resulting in the cerebellar granule neurons incapable of being induced to repetitively fire AP (Reference Goldfarb, Schoorlemmer and Williams18). In addition, a mutation of the NaV1.7 channel alters the excitability in different types of neurons (Reference Rush, Dib-Hajj, Liu, Cummins, Black and Waxman19). In the sensory neurons of the PNS, the Na+ channels contribute to inflammatory and neuropathic pain (Reference Hains, Saab and Klein20), and serve as analgesic drug targets (Reference Momin and Wood21).
Despite the evidence that both IL-6 and voltage-gated sodium channels are involved in neuronal pathogenesis and protection, there is still limited information on the role of IL-6 in voltage-gated sodium channels. In the present study, we focused for the first time on voltage-gated sodium channels to indicate the mechanism of the neuroprotective effect of IL-6 on voltage-gated sodium channels by means of whole-cell patch-clamp methods and real-time polymerase chain reaction (PCR).
Materials and methods
Spinal cord neuron culture
The primary culture of rat spinal cord neurons was performed according to the previous reports (Reference Momin and Wood21,22). The animals used in this study were housed and treated according to the guidelines for care and use of experimental animals of the Ethics Committee of Xiangya University Medical College. Primary cultures of spinal cord neurons were prepared from embryonic day 14 (E14) foetal rats. Sprague–Dawley rats were purchased from Central South University Xiangya Medical College Experimental Animal Centre, Changsha, China. All procedures were measured up to the national standard. Briefly, the spinal cord cleaned of its meningeal membranes was trypsinised for 2 min with 4 ml of 0.25% trypsin (Invitrogen, NY, USA) at 37°C, and interrupted with a 0.5 ml foetal bovine serum (FBS, HyClone). The cells were collected by centrifugation for 10 min at 900× g. The pellet was resuspended by minimum essential medium (MEM, Invitrogen). 1 × 106 cells were plated onto a 22 × 22 mm glass cover slip that was pretreated with 12.5 μg/ml Poly-d-Lysine (Sigma; in water). 2 mM glutamine (Sigma), 1% B-27 supplement and 10% FBS were added into the MEM immediately before use. The 35 mm dishes were incubated at 37°C, 5% CO2/95% air with 2 ml culture medium. Half of the culture medium was changed every 3 days. All experiments were conducted at days 6–12 in vitro.
A different dose of IL-6 (Sigma) was applied to the medium for 24 h, or 10 ng/ml of IL-6 was applied for 4, 8, 24 or 48 h. For pharmacological experiments, IL-6 receptor antibody (1 μg/ml; Sigma) and/or anti-gp130 antibody (1 μM; Sigma) were applied to medium at 30 min before IL-6 (10 ng/ml) and then the cultures was co-treated for 24 h.
Electrophysiological recording
Electrophysiological recording of the rat spinal cord neurons was carried out according to the previous reports (Reference Zhou, Qi and Zhao17). For the whole-cell recording of the voltage-gated Na+ currents, the bath solution had the following composition (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 Glucose and pH 7.4 with NaOH; the pipette solution contained (in mM): 145 CsCl, 1 MgCl2, 10 EGTA, 4 TEA-Cl, 10 HEPES, 5 ATP-Na2 and pH 7.2 with CsOH. Patch pipettes were pulled to a tip resistance of 5–10 MΩ from a borosilicate glass capillary tube on a two-stage puller (PP83; Narishige, Tokyo, Japan). Voltage-clamp recording was obtained using an Axon-200B amplifier (Molecular Devices Corporation, Sunnyvale, USA). The series resistance was compensated by 50–70%. Data were digitised at 200 kHz. To examine the activation properties, neurons were held at −80 mV and depolarised to potentials ranging from −80 to 60 mV for 20 ms with 10 mV stepping and 0.5 Hz frequency. To examine the inactivation properties, Na+ currents were recorded at −25 mV after pre-pulse from −100 to 0 mV for 40 ms with 10 mV stepping.
To measure the amplitude and threshold of the spike, current-clamp recording was carried out in the above bath solution. The pipette solution contained (in mM): 130 KCl, 10 NaCl, 1 MgCl2, 10 EGTA, 10 HEPES, 10 glucose, 5 ATP-Na2 and pH 7.2 with KOH. The neurons were held around −70 mV by injecting a hyperpolarisation current, and the spike was elicited by a 500 pA depolarising ramp current for 100 ms. All experiments were conducted at room temperature.
Total RNA extraction, reverse transcription and real-time PCR
To determine the effects of IL-6 on the expression of Na+ channel proteins, the content of α1A subunit mRNA was measured using real-time PCR. For this experiment, to maximise the percentage of the neurons, the cells were cultured serum-free in Neurobasal-A medium (Invitrogen) with 2% B-27. A selective inhibitor of DNA synthesis, arabinosylcytosine C (10 μM), was added to the medium at day 3 for 24 h to eliminate the glia cells, such as astrocytes.
Total RNA was extracted from cultures using TRIZOL (Invitrogen) according to the manufacturer's instructions. The purified RNA was reverse transcribed to cDNA with oligo dT-20 primers. Real-time PCR was performed using Mx3000p QPCR system (Stratagene Corporation, USA) with SYBR green fluorophore. A total reaction volume of 20 μl contained 10 μl SYBR Green PCR Master Mix (Applied Biosystems, NY, USA), 1 μl cDNA template and 0.8 μl forward/reverse primers (SCN1α forward: 5′-GATGTTCTACGAGGTCTGG-3′; SCN1α reverse: 5′-AGTTTGTTCGGTTGT GGT-3′0; GAPDH forward: 5′-ATCAAGAAGGTGGTGAAGCA-3′; GAPDH reverse: 5′-AAGGTG GAAGAATGGGAGTTG-3′). The templates were amplified using the following PCR protocol: 95°C for 10 min, 40 cycles of 95°C for 30 s and 60°C for 1 min.
Immunoblot analysis
Membranes were purified by discontinuous sucrose gradient centrifugation. Briefly, whole-brain lysates in 0.32 M sucrose/5 Mm Tris, pH 7.4 were layered onto 1.2 M sucrose/5 mM Tris, pH 7.4 and centrifuged at 100 000 g for 30 min. The protein fraction at the 0.8–1.2 M sucrose interface was collected, diluted twofold with 0.8 M sucrose/5 mM Tris, pH 7.4 and centrifuged at 20 000 g for 20 min. Pelleted membrane proteins were resuspended in one RIA buffer (25 mM Tris, 150 mM NaCl, 1 mM EDTA, 2% Triton X-100, pH 7.4) and centrifuged at 20 000 g for 20 min, and the supernatant containing the membrane proteins was collected for analysis. Complete Protease Inhibitor set (Roche, Basle, Switzerland) was included in all solutions. Membrane proteins (100 mg) were fractionated by SDS-PAGE and immunoblotted using specific sodium channel antibodies: anti-Na V1.1 (Abcam; diluted 1 : 500).
Data analysis
Data were expressed as mean ± SEM. Statistical analysis was performed using Origin software (Origin Lab Corporation, Northampton, USA) and SPSS 15.0 (SPSS Inc., USA). One-way ANOVA followed by LSD was used for comparison of the normative Na+ currents amplitudes, current density (pA/pF), currents dose-dependent and time-dependent, the thresholds and ampulitudes of spikes, qPCR data and protein expression level. Student's t-test was used for the comparison of activation curve and inactivation curve between two groups. For real-time PCR, the results were analysed with the Sequence Detector Software version 2.1 (Applied Biosystems). According to the comparative threshold cycle (CT) method, the relative gene expression was calculated by the formula 2−(ΔΔCT).
Results
Dose-dependence of the inhibition of IL-6 on Na+ currents
The spinal cord cultures were treated with IL-6 at 0.1, 1 and 10 ng/ml for 24 h before the whole-cell voltage-gated Na+ currents were recorded. An inward current with fast activation and inactivation was elicited by depolarisation from −80 mV of the holding potential and recorded at 200 kHz of the sampling frequency (Fig. 1a). To obviate the difference in Na+ currents caused by the various sizes of the neurons in our recording, the current density was calculated by the peak current divided by membrane capacitance that was obtained in the lock-in simulative protocol of the Axon-200B amplifier. The voltage-gated Na+ currents were significantly inhibited at the most activated potentials by 1 ng/ml or higher (10 ng/ml) of IL-6, but not by 0.1 ng/ml of IL-6 (Fig. 1b). The peak currents, up to −10 mV, were plotted to the dosages of IL-6 and the results showed that the inhibition by IL-6 appeared to be dose-dependent. The Na+ currents were suppressed by ∼6, 39 and 46% by 0.1, 1 and 10 ng/ml of IL-6, respectively (Fig. 1c). IL-6 receptor antibody was used to test whether the IL-6 inhibition is IL-6R-dependent, and the result showed that IL-6ra abolished the inhibitory effects of IL-6 on Na+ currents and did not alter the currents when used alone (Fig. 1d). Anti-gp130 antibody at 1 μg/ml was used to test whether the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signal transduction pathway contribute to IL-6 inhibition and subsequent result showed that anti-gp130 antibody abolished the inhibitory effects of IL-6 on Na+ currents (Fig. 1e).

Fig. 1 The effects of IL-6 on voltage-gated Na+ currents were dose-dependent and through its receptor and gp130 in the cultured spinal cord neurons of rats. (a) A typical recording curve of whole-cell Na+ currents from −80 to 60 mV in the control and IL-6-treated (10 ng/ml, 24 h) neurons. (b) The current density–voltage relationship in 0.1 and 10 ng/ml (24 h) of IL-6-treated groups. *p < 0.05 and **p < 0.01 as compared with control (one-way ANOVA followed by LSD test). (c) The dose-dependency of IL-6 effects on Na+ currents ranged from 0.1 to 10 ng/ml. The current data were normalised by control respectively and expressed as percentage of control group. (d) The normative Na+ currents amplitudes at −10 mV in control, IL-6 only, IL-6+IL-6ra and IL-6ra only groups. (e) The normative Na+ currents amplitudes at −10 mV in control, IL-6 only, IL-6+anti-gp130 antibody; anti-gp130 antibody only *p < 0.05 and **p < 0.01 as compared with control; #p < 0.05 and ##p < 0.01 as compared with IL-6 group (one-way ANOVA followed by LSD test).
Time-dependence of IL-6 inhibition on Na+ currents
To examine whether the effects of IL-6 on Na+ currents are time-dependent, 10 ng/ml of IL-6 was chosen in the further experiments. The neurons were treated with IL-6 at 10n g/ml for 4, 8, 24 and 48 h, respectively, and results showed that IL-6 inhibited the voltage-gated Na+ current significantly at 24 h treatment (Fig. 2a). The reduction of the currents induced by IL-6 at, 4, 8, 24 and 48 h were 28%, 42%, 51% and 22%, respectively (Fig. 2b).
Fig. 2 The effects of IL-6 on voltage-gated Na+ currents were time-dependent in the cultured spinal cord neurons of rat. (a) The current density–voltage relationship in 10 ng/ml of IL-6-treated for 24 and 48 h, respectively. *p < 0.05 and **p < 0.01 as compared with control (one-way ANOVA followed by LSD test). (b) The time-dependency of IL-6 (10 ng/ml) effects on Na+ currents ranged from 4 to 48 h. The current data were normalised by control respectively and expressed as percentage of control group. *p < 0.05 and **p < 0.01 as compared with control (one-way ANOVA followed by LSD test).
Effects of IL-6 on the activation and inactivation of Na+ channels
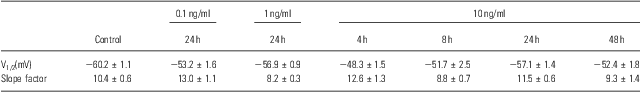
To identify other possible effects of IL-6 on the voltage-gated Na+ channels, the voltage-dependent activation and inactivation properties of the Na+ channels at each dose and period of IL-6 treatments were analysed and compared. For the activation, the conductance at each voltage was calculated by the peak current and driving potential and then normalised by the maximal conductance. The normative conductance was plotted to the depolarised voltage, and the conductance–voltage curves were fitted using the Boltzmann equation (Fig. 3a). For the inactivation analysis, the peak currents at −25 mV of the test voltage were normalised by the maximal current (Fig. 3b). The normative currents were plotted to the prepulse voltage, and the current–voltage curves were also fitted using the Boltzmann equation (Fig. 3b). Neither the activation nor inactivation properties were affected by IL-6 at any doses or administered time used in this study – even those that significantly reduced the whole-cell Na+ currents (e.g. 10 ng/ml for 24 h; Fig. 3a and b). The entire parameters (i.e. V1/2 and slope factor) of voltage-dependent activation and inactivation were summarised in Tables 1 and 2.
Fig. 3 The effects of IL-6 on electrophysiological characteristics. (a) Voltage-dependent activation curves (Boltzmann equation and fitting parameters) in 10 ng/ml treated for 24 h and control groups. (b) A typical curve for the analysis of inactivation properties (insert) and voltage-dependent inactivation curves (Boltzmann equation and fitting parameters) in 10 ng/ml treating for 24 h and control groups. *p < 0.05 as compared with control (Student's t-test).
Table 1 Parameters of voltage-dependent activation
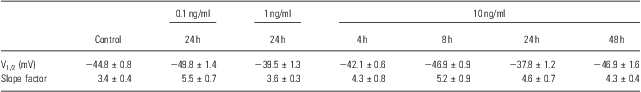
Table 2 Parameters of voltage-dependent inactivation
Using real-time PCR, we estimated α1A (SCN1α) subunit mRNA in the neurons and found that the α1A subunit expression was significantly inhibited by IL-6 at 10 ng/ml for 2 h. This expression inhibition was transient, peaked at 2 h and recovered at 4 h and thereafter (Fig. 4a). The result of the immunoblot analysis showed that SCN1α subunit protein level was suppressed steadily by IL-6 (10 ng/ml) at 4, 8 and 24 h (Fig. 4b and c). Together these results suggest that IL-6 inhibits the Na+ currents through not changing the essential characteristics of the voltage-gated Na+ channels in the neurons, but reducing the amount of channels in the plasma membrane.
Fig. 4 The expression of α1A subunit in mRNA level and protein level with control. (a) The expression of α1A subunit was significantly reduced by IL-6(10 ng/ml) treatment at 2 h, but not 4, 8 or 24 h. *p<0.05 as compared with control; #p < 0.05 and ##p < 0.01 as compared with 2 h group (one-way ANOVA followed by LSD test). (b) The protein expression of the SCN1α subunit was significantly downregulated by IL-6 (10 ng/ml) at 4, 8 and 24 h. (c) The data are from three separate experiments. *p < 0.05, compared with control (one-way ANOVA followed by LSD test).
Effects of IL-6 on amplitude and threshold of the spike
Voltage-gated Na+ channels are essential in achieving the rising phase and overshoot of the AP. To probe the IL-6 effects on the AP, the spikes were recorded in a current-clamp configuration (Fig. 5). The threshold of the spike was not affected by IL-6 (Fig. 5c). On the other hand, the peak of the spike was lowered significantly by 10 ng/ml of IL-6 for 24 h, but not changed at 1 or 0.1 ng/ml IL-6 (Fig. 5d). These results agree well with the data from the current density recording and suggest that IL-6 reduces the amplitude of the AP by inhibition on the voltage-gated Na+ channels in the cortical neurons.
Fig. 5 The effects of IL-6 on spike. (a and b) Typical curves of spike elicited by a ramp depolarised current in control group (a) and IL-6 (10 ng for 24 h) treated group (b). (c and d) The thresholds (c) and amplitudes (d) of spike in control (n = 31), 0.1 (n = 26), 1 (n = 21) and 10 (n = 18) ng/ml IL-6-treated groups. *p < 0.05 as compared with control (one-way ANOVA followed by LSD test).
Discussion
IL-6 plays an important role in various pathophysiological conditions of the CNS via its receptor-mediated intracellular signal cascades (Reference Nelson, Ur and Gruol23). Some studies have reported on the relationship between IL-6 and ion channels in the neurons, including acute effects on l-type calcium channel of the cultured cerebellar granule neurons (Reference Ma, Li and Huang24), chronic effects on the mean amplitude of the calcium signal in response to glutamate receptor agonists in the cultured cerebellar Purkinje neurons (Reference Nelson, Ur and Gruol23). This study newly found that IL-6 can inhibit the voltage-gated Na+ currents, but not voltage-dependent activation/inactivation (e.g. AP) in the cultured spinal cord neurons. It is known that the reduction of Na+ currents declines energy consumption that is used for maintaining the Na+ gradient across the plasma membrane in the injured neurons. Thereby, IL-6 suppressed Na+ currents in the spinal cord neurons may promote neuronal survival in injuries and diseases of the CNS. On the other hand, an extensive amount of literature suggests that IL-6 has a negative effect on neuronal survival and neuroprotection in an in vivo model of CNS disease (Reference Spooren, Kolmus and Laureys25–Reference Allan and Rothwell27). Both real-time PCR and immunoblot results revealed the decrease of the channel gene expression, and the antibody of IL-6 receptor abolished this inhibition, indicating that IL-6 functions through its receptor-mediated mechanism.
The characteristics of IL-6-induced changes in the physiological properties of the spinal cord neurons were dependent on the dose of IL-6 and the duration of exposure. However, the relationship between the dose/duration of IL-6 and the effect was not always straightforward. For example, 0.1–10 ng/ml IL-6 exposure for 24 h resulted in dose-dependent reductions in Na+ currents, whereas at 10 ng/ml of IL-6, Na+ currents were decreased significantly at 4, 8 and 24 h. The currents returned nearly to the control level after 48 h. Similar to the voltage data, the mRNA level of α1A subunit was reduced at 2 h but recovered sooner. These results implicate that IL-6 regulates the Na+ channel activity by reducing its expression. The level of mRNA returns to normal at 4 h, whereas currents were reinstated at 48 h, which indicates that the switch-on of Na+ channel gene expression is an immediate event with a narrow window, but the recovery of currents may depend on the half-life of the proteins. Further approach is warranted to clarify this question.
It has been reported that IL-6 acts on the target cells and promotes dimerisation of gp130, a signal-transducing subunit coupled with the IL-6 receptor (Reference Nelson, Ur and Gruol23,Reference Peng, Qiu, Lu and Wang28), which is confirmed in our experiments (Fig. 1b and c). The spinal cord neurons express IL-6 receptors and gp130 signal protein (Reference Yang, Wen and Ou29,Reference Kang30). In this study, anti-gp130 antibody blocked the inhibitory activity of IL-6 on glutamate-induced intracellular Ca2+ overload, indicating that the IL-6 receptor is involved in the neuroprotective effect of IL-6. Therefore, we deduce that the IL-6-induced inhibition of voltage-gated sodium activities is achieved through a receptor-mediated mechanism.
A spike was recorded using current-clamp technique in our experimental conditions. The threshold of the spike was stable in various experimental groups, in agreement with the stability of the voltage-dependent activation of the Na+ currents. The current-clamp recording data showed that IL-6 decreased the AP amplitude of neurons, in which Na+ currents were also inhibited. A large number of studies have indicated that IL-6 can reduce neuronal death via a reduction of Ca2+ entry into the neurons (Reference Nelson, Ur and Gruol23,Reference Georgene, Anthony and J Philip31). The reduction of AP amplitude may indirectly decrease the Ca2+ influx through voltage-gated Ca2+ channels during the spike, and thus protect the neurons from injury induced by Ca2+ overload. Excitotoxicity resulting from the excessive release of excitatory transmitters is involved in many CNS diseases and injuries (Reference Jabaudon, Scanziani, Gaähwiler and Gerber32,Reference Nishizawa33). The reduction of AP amplitude revealed that IL-6 may decrease the release of neurotransmitters from the presynaptic terminal.
In conclusion, we demonstrated that IL-6 inhibits the voltage-gated Na+ currents in a time- and dose-dependent manner through IL-6 receptor and gp130, but does not affect the voltage-dependent activation and inactivation in the spinal cord neurons. These results suggest that IL-6 may regulate the properties of the AP in the neurons via its effects on Na+ channels. The potential neuroprotection of IL-6 may be ascribed to its inhibitory effect on Na+ currents, reducing energy consumption in the injured neurons or the release of excitatory transmitters in excitotoxicity.
Acknowledgement
This work was supported by the National Natural Science Foundation of China (No. 30800572).




 Open access
Open access