



As a crucial type of layered material, phyllosilicate minerals have played a significant role in human life and civilization for a long time (Zhang et al., Reference Zhang, Lin, Lv, Xiao, He, Wang and Li2014). Pyrophyllite is one of the most abundant phyllosilicate minerals (Du & Yuan, Reference Du and Yuan2019). Because of its beneficial physicochemical and mechanical properties (Zhao et al., Reference Zhao, Cao, Wang and Zhang2021a), pyrophyllite is used in construction materials, ceramics, refractories, chemicals, the paper and coating industries and other fields (Bentayeb et al., Reference Bentayeb, Amouric, Olives, Dekayir and Nadiri2003). Therefore, many researchers have investigated the physicochemical and mechanical properties of pyrophyllite using experimental and theoretical computation methods (Refson et al., Reference Refson, Park and Sposito2003; Katti et al., Reference Katti, Schmidt, Ghosh and Katti2005; Shi et al., Reference Shi, Liu, Xin, Lin, Zhao and Liu2017), increasing our understanding of pyrophyllite, which is now considered viable for use in coatings, medicines, cosmetics, the aerospace industry and other fields (Ali et al., Reference Ali, Ahmed, Ahmed and Hefni2021; Luna et al., Reference Luna, Reimers, Avena and Juan2021).
However, natural pyrophyllite includes a wide range of defect elements. Yan et al. (Reference Yan, Zhang, Fang, Dong, Shao and Sheng2013) measured the mass fractions of Na2O, MgO, K2O and Fe2O3 in pyrophyllite samples from Zhejiang Province, China, and found them to be 0.27, 0.13, 0.67 and 0.45 wt.%, respectively. Reddy et al. (Reference Reddy, Reddy and Endo2016) determined the mass fractions of Na2O, MgO, K2O, Fe2O3 and CaO in pyrophyllite minerals from California at 0.10, 0.01, 0.20, 0.20 and 0.09 wt.%, respectively. Based on above results and other data (Yan et al., Reference Yan, Zhang, Fang, Dong, Shao and Sheng2013; Reddy et al., Reference Reddy, Reddy and Endo2016; Shi et al., Reference Shi, Liu, Xin, Lin, Zhao and Liu2017), the primary contaminant defect elements in pyrophyllite are Na(I), K(I), Mg(II), Ca(II) and Fe(II), with concentration ranges of 0.04–0.27, 0.02–0.35, 0.01–0.47, 0.02–0.31 and 0.07–0.54 wt.%, respectively.
As is well known, metal cations often enter pyrophyllite crystals via cation substitution (Zhao, Reference Zhao2013). The differences in the sizes and charges of the ions that substituted into pyrophyllite affect the atomic and electronic structures of pyrophyllite, leading to changes in the physicochemical and mechanical properties of pyrophyllite (Zhao, Reference Zhao2013). Ali et al. (Reference Ali, Ahmed, Ahmed and Hefni2021) experimented with minor iron doping into pyrophyllite, which resulted in a reduction in the melting point of refractories, influenced the transparency of glass products and decreased the transmission capabilities of optical fibres. Luna et al. (Reference Luna, Reimers, Avena and Juan2021) highlighted the impacts on the work function, magnetic moment and other parameters when doping pyrophyllite with Mg(II), Fe(II) and Fe(III). Based on the above experimental results, the primary contaminant defect elements of pyrophyllite are Na(I), K(I), Mg(II), Ca(II) and Fe(II). To the best of the authors’ knowledge, the effects of doping using these five elements on the electronic structure and mechanical properties of pyrophyllite have not been established. Greater insights into the doping effects of Na(I), K(I), Mg(II), Ca(II) and Fe(II) on the various properties of pyrophyllite are needed, which could be obtained through detailed first-principles calculations at the molecular level.
In this work, the doping formation mechanism and influences of Na(I), K(I), Mg(II), Ca(II) and Fe(II) doping on the atomic and mechanical characteristics of pyrophyllite were calculated using density functional theory (DFT). By comparing the lattice parameters, the partial density of state (PDOS), total density of state (TDOS), the band structure and the elastic constants of pyrophyllite with various doping agents, the effects of doping on these physicochemical and mechanical properties can be obtained. The objective of this study was to use a first-principles approach to obtain an understanding of the various properties associated with various point defects in pyrophyllite at the molecular level.
Materials and methods
The Vienna Ab initio Simulation Package (VASP; Kresse & Furthmüller, Reference Kresse and Furthmüller1996; Tunega et al., Reference Tunega, Bŭcko and Zaoui2012) was applied to pyrophyllite with the molecular formula Al4Si8O24H4 and space group P1. The calculation of the exchange correlation energy was performed using the Perdew–Burke–Ernzerhof (PBE) density functional (Perdew et al., Reference Perdew, Burke and Ernzerhof1996; Putra et al., Reference Putra, Muttaqien, Hamamoto, Inagaki and Morikawa2019). Through the utilization of calculated Hellmann–Feynman forces, a comprehensive relaxation of all atomic positions in pyrophyllite was performed. The energy cutoff for the plane-wave basis was 600 eV. To optimize the atomic geometries, a conjugate-gradient algorithm was employed, refining the positions of the atoms iteratively. The optimization process continued until the residual force acting on the atoms reached a threshold of <0.01 eV Å–1 (Zhao et al., Reference Zhao, Qin, Wang and He2020). The 9 × 5 × 5 Monkhorst–Pack (Chen et al., Reference Chen, Xie, Zhang and Sun2020; Hou et al., Reference Hou, Chen, Zhou, Bian, Dong and Tang2020; Zhao et al., Reference Zhao, Cao, Wang and Zhang2021a) k-points set was used. The valence electrons of pyrophyllite included 1s 1 of H, 2s 2 and 2p 4 of O, 3s 2 and 3p 1 of Al and 3s 2 and 3p 2 of Si. The valence electrons of doped atoms in the pyrophyllite included 3p 63d 64s 2 of Fe, 2p 63s 2 of Mg, 2p 63s 1 of Na, 2p 63s 23p 64s 1 of K and 2p 63s 23p 64s 2 of Ca.
All of the calculations were performed using a 1 × 1 × 1 unit cell (containing 40 atoms) as shown in Fig. 1. The defect system was modelled by arranging for a defect to substitute for Al atoms of a periodic unit cell (Fig. 1a). The 1 × 1 × 1 unit cell was chosen to provide sufficient lattice sites to accommodate defect concentrations as low as 0.037–0.149 wt.%, similar to previous studies. First, the doping mechanism of various metal defect substitutions of the pyrophyllite Al ion was calculated (Fig. 1a). To determine the doping mechanism of various metal defects in the pyrophyllite, the formation energy Ef(X, q) was calculated, given by Equation 1 (Walle & Neugebauer, Reference Walle and Neugebauer2004):

where E tot(X,q) and E (pyrophyllite) are the energies of the doped and defect-free pyrophyllite systems, respectively; ni and nX are the numbers of Al (Si, H or O) and Na (K, Mg, Ca or Fe) atoms of the unit cell, respectively; and ui and uX represent the chemical potentials of Al (Si, H or O) and Na (K, Mg, Ca or Fe) atoms, respectively. E F represents the Fermi energy measured from the valence band maximum E V of the undoped pyrophyllite, and ΔV is a correction term for aligning the electrostatic potential to E V.
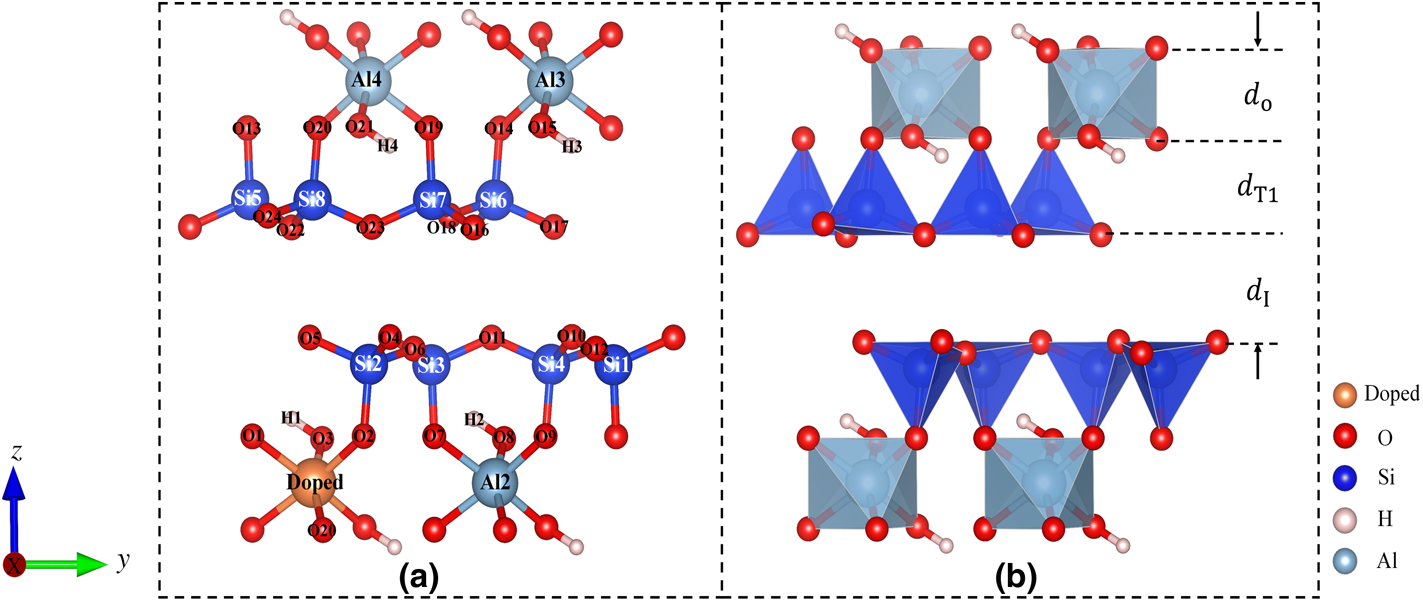
Figure 1. (a) Unit cell of pyrophyllite (Al4Si8O24H4) including 40 atoms. The orange sphere represents the Al ion replaced by a defect. (b) A layer of 2:1 pyrophyllite marked with layer distances.
Table 1 presents the formation energy results of five kinds of defects in the pyrophyllite. The formation energies of the Na(I), K(I), Mg(II), Ca(II) and Fe(II) doping were 4.886, 5.229, 3.492, 3.810 and 1.165 eV, respectively, denoting that heat needed to be absorbed from outside during doping. The formation energy of Fe(II)-doped pyrophyllite was the lowest, with the other formation energies following the order of Mg(II) < Ca(II) < Na(I) < K(I). These results indicate that the stability of the Fe(II)-doped pyrophyllite was greater than those of the other doped pyrophyllites, and the Fe atoms were substituted for the Al atoms of the pyrophyllite more easily than the other doping ions.
Table 1. Total energy of the pyrophyllite system with various types of dopant atom and the formation energies of the dopant atoms.

Based on the above optimized structure of the pyrophyllite with metal doping, other properties were investigated further. By subjecting the equilibrium lattice to stress tensors with small strains, the elastic constants Cij of the crystalline systems were determined based on the corresponding changes in the total energy of the unit cell. In this work, the strain amplitude δ was varied with steps of 0.01 from –0.06 to 0.06 (Qin et al., Reference Qin, Zhao, Wang and He2020). The bulk modulus (B; Equation 2) and shear modulus (G; Equation 3) of pyrophyllite can be calculated using the Voigt–Reuss–Hill (VRH) approximation based on the elastic constants obtained (Hill, Reference Hill1952; Chung & Buessem, Reference Chung and Buessem1968):


For a detailed computation of B R, B V, G R and G V, see Qin et al. (Reference Qin, Zhao, Wang and He2020). Young's modulus (Y), Poisson's ratio (μ), acoustic compression (υp) wave velocity and shear (υs) wave velocity were obtained using Equations 4–7.




where ρ represents the density of the pyrophyllite.
Results and discussion
Effects on the atomic structure of pyrophyllite
Pyrophyllite, with the ideal structural formula Al4Si8O24H4, is a planar 2:1 phyllosilicate composed of two-dimensional mineral layers. Existing experimental data (Gruner, Reference Gruner1934; Lee & Guggenheim, Reference Lee and Guggenheim1981) and previously calculated results (Bruno et al., Reference Bruno, Prencipe and Valdre’2006; Kremleva et al., Reference Kremleva, Martorell, Kruger and Rosch2012; Lavikainen et al., Reference Lavikainen, Hirvi, Kasa, Schatz and Pakkanen2015) regarding the pyrophyllite layers provided the data basis for the establishment of the structure of pyrophyllite (Zhang et al., Reference Zhang, Wei, Ferrell, Guggenheim, Cygan and Luo2010). In our first-principles calculations, an atomic structure was determined to represent the positions of atoms and lattice parameters after optimization. Table 2 shows the lattice parameters of undoped pyrophyllite and Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllite. The calculated structural parameters of undoped pyrophyllite were consistent with previous experimental data (Gruner, Reference Gruner1934; Lee & Guggenheim, Reference Lee and Guggenheim1981). Compared to the lattice parameters of undoped pyrophyllite and doped pyrophyllite, as listed in Table 2, the effects of K(I) doping on pyrophyllite were the greatest, and the least effects were observed for Fe(II) doping of pyrophyllite.
Table 2. The lattice parameters of the undoped and doped pyrophyllites.

Table 3 shows the average calculated bond lengths of the undoped and doped pyrophyllites. The H–OH bond length of doped pyrophyllite was longer than that of undoped pyrophyllite. In addition, the average Al2–OH bond length of doped pyrophyllite was longer than that of undoped pyrophyllite, whereas the Al2–Oa bond length of doped pyrophyllite was shorter than that of undoped pyrophyllite (Putra et al., Reference Putra, Muttaqien, Hamamoto, Inagaki and Morikawa2019). The average Si2–Oa and Si2–Ob bond lengths of doped pyrophyllite were shorter than those of undoped pyrophyllite. Finally, the orders of bond lengths of doping atoms with oxygen atoms were K–OH > Ca–OH > Na–OH > Mg–OH > Fe–OH > Al2–OH and K–Oa > Ca–Oa > Na–Oa > Mg–Oa > Fe–Oa > Al2–Oa.
Table 3. The calculated average bond lengths of the undoped and doped pyrophyllites.

As shown in Fig. 1b, the changes of layer thickness before and after doping of pyrophyllite were also calculated (Table 4), where d I was the thickness of the two-dimensional layer, d T1 was the thickness of the tetrahedral (SiO4) sheet, and d o was the thickness of the octahedral (AlO6) sheet (Qin et al., Reference Qin, Zhao, Wang and He2020). Compared with the values of the undoped pyrophyllite, the d o values of Na(I)-, K(I)-, Mg(II)- and Ca(II)-doped pyrophyllite increased by 0.43%, 6.69%, 0.05% and 2.09%, respectively, whereas the value of Fe(II)-doped pyrophyllite decreased by 1.23%. Moreover, the d T1 values of Na(I)-, Mg(I)-, Ca(II)- and Fe(II)-doped pyrophyllite were greater than that of the undoped pyrophyllite by 0.32%, 1.75%, 0.32% and 1.30%, respectively, whereas the d T1 value of K(I)-doped pyrophyllite was less than that of undoped pyrophyllite by 0.69%. Finally, the interlayer thickness d I of the doped pyrophyllite was reduced compared with that of the undoped pyrophyllite. All of the above results demonstrate that doping has an influence on the lattice parameters, interlayer thickness and bond lengths of pyrophyllite.
Table 4. The average layer thicknesses and interlayer thicknesses of the undoped and doped pyrophyllites.

Effects on the electronic properties of pyrophyllite
The PDOS, TDOS and band structure of the undoped and doped pyrophyllites were calculated to determine the effects of defects on the electronic properties of pyrophyllite in detail (Tunega et al., Reference Tunega, Bŭcko and Zaoui2012).
Figure 2 presents the band structure of pyrophyllite along the high-symmetry lines of the Brillouin zone (BZ). G(0, 0, 0), F(0, 0.5, 0), Q(0, 0.5, 0.5) and Z(0, 0, 0.5) were the high-symmetry BZ points of pyrophyllite (Zhao et al., Reference Zhao, Wang, Gao, Wang and Huang2021b; Wang et al., Reference Wang, Zhao, Qiao and Luan2022). The electronic energy band structure of the undoped pyrophyllite was 5.54 eV, as shown in Fig. 2a. The Z point was the valence band maximum (VBM) of the undoped pyrophyllite, while the G point was the conduction band minimum (CBM). Figure 2b–f shows the band structure of Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllite, with gap widths of 5.65, 5.33, 5.59, 5.54 and 2.04 eV, respectively. The VBMs of Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllite were the Q, G, Q, Z and Q points, respectively, while the CBMs of all of the doped pyrophyllite band structures were at the G point. These results show that the effects of Fe(II) doping on band structure of pyrophyllite were the greatest, while the least effects were observed for Ca(II) doping of pyrophyllite. All of the undoped and doped pyrophyllites remained as insulators, which is one of the reasons why pyrophyllite can be used as a pressure transmission medium and in dielectric ceramics and other fields.

Figure 2. The band structures of the (a) undoped, (b) Na-doped, (c) K-doped, (d) Mg-doped, (e) Ca-doped and (f) Fe-doped pyrophyllites.
The PDOS and TDOS of undoped and doped pyrophyllites are depicted in Fig. 3a–f. The Fermi energy level was set as the reference point and assigned a value of 0. The PDOSs of O1, O2 and O3 were plotted in Fig. 3 separately because these three kinds of oxygen atoms have different symmetries and positions in the unit cell. The PDOSs of these three atoms were similar to each other. A large charge transfer to the O 2p states from the Al 3p, Na 2p, K 3p, Mg 2p, Ca 3p and Fe 3d states occurred because of the high electronegativity of oxygen. This phenomenon led to the observed similarity. As shown in Fig. 3a, in the wide energy range of 10 eV < E < E F, the valence bands primarily consisted of O 2p states. Furthermore, some residual charges were also to be found in the Al 3s/3p and Si 3s/3p states. The covalent components of the Al–O and Si–O chemical bonds of pyrophyllite were observed. Compared with the PDOS of Al atoms, Mg(II) doping of pyrophyllite led to similar valence bands in the energy range 10 eV < E < E F, composed of O 2p states. Doping of Na 2p, K 3p and Ca 3p states transferred fewer electrons to O 2p states than Al 3p states. After Fe(II) doping of pyrophyllite, the PDOS of the O atoms changed significantly, and strong hybridization between the O p and Fe d orbitals occurred because the O atoms presented new peaks and aligned with the bonding orbital of Fe(II). This phenomenon was due to the charge redistribution resulting from the varying electronegativities of the doping elements and O atoms. This redistribution led to a global electrostatic attraction between the O atoms and Na(I), K(I), Mg(II), Ca(II) and Fe(II) after doping of pyrophyllite (Zhao & He, Reference Zhao and He2014).

Figure 3. The total and partial densities of states of the (a) undoped, (b) Na-doped, (c) K-doped, (d) Mg-doped, (e) Ca-doped and (f) Fe-doped pyrophyllites.
Effects on the mechanical properties of pyrophyllite
As is well known, the mechanical properties of a material are crucial in engineering, industry and other applications. Elastic properties represent the innate properties of a material (Zhao & He, Reference Zhao and He2014) that indicate the degree to which it can deform under external stress (Pawley et al., Reference Pawley, Clark and Chinnery2002). The mechanical properties of undoped and doped pyrophyllites were investigated systematically. The elastic stiffness constants (Cij) of a crystalline system are essential parameters that describe the mechanical properties of its materials, such as its bulk modulus, Young's modulus, etc. (Yang et al., Reference Yang, Wang, Gan, Shi and Tang2018). Pyrophyllite is a triclinic system with 21 independent elastic stiffness constants (Benazzouz & Zaoui, Reference Benazzouz and Zaoui2012). C 11, C 22, C 33, C 44, C 55, C 66, C 12, C 13, C 14, C 15, C 16, C 23, C 24, C 25, C 26, C 34, C 35, C 36, C 45, C 46 and C 56 were calculated (Zhao et al., Reference Zhao, Qin, Wang and He2020). The results for the undoped and doped pyrophyllites are listed in Table 4. First, the results of the present first-principles calculation for the undoped pyrophyllite were similar to previous experimental data (Cheng et al., Reference Cheng, Sondergeld and Rai2013) and other previously calculated results (Zartman et al., Reference Zartman, Liu, Akdim, Pachter and Heinz2010; Li, Reference Li2016), meaning that the present computational approach is highly reliable. Furthermore, it was found that the Born–Huang criteria (Huang et al., Reference Huang, Jiang, Yang, Xiong, He and Zhu2019) were satisfied for all Cij of the undoped and doped pyrophyllites, indicating that the triclinic undoped and doped pyrophyllites were mechanically stable.
Finally, the calculated longitudinal classic constants of the undoped pyrophyllite were C 11 = 183.19 GPa, C 22 = 196.92 GPa and C 33 = 51.58 GPa, as shown in Table 4. The elastic constant C 11 was less than C 22, indicating that the anti-deformation ability of the b-axis was stronger than that of the a-axis. Consistent with expectations, the crystal exhibited the greatest susceptibility to deformation along the c-axis, as represented by the elastic constant C 33. In addition, the elastic constant C 66 was greater than C 44 and C 55, indicating that the shear deformation resistances of the (100) and (010) planes were weaker than that of the (001) plane. It was found that C 11, C 22, C 33 and C 66 of doped pyrophyllite were less than those of undoped pyrophyllite. In particular, C 11 of the K(I)-doped pyrophyllite decreased the most (by 28.02%), C 22 of the K(I)-doped pyrophyllite decreased the most (by 31.64%), C 33 of the Na(I)-doped pyrophyllite decreased the most (by 20.32%) and C 66 of the K(I)-doped pyrophyllite decreased the most (by 25.72%). However, C 44 of Na(I), Mg(II), Ca(II) and Fe(II)-doped pyrophyllite were greater than those of undoped pyrophyllite, and C 44 of Fe(II)-doped pyrophyllite was less than that of undoped pyrophyllite. In addition, C 55 of Mg(II)-, Na(I)- and Ca(II)-doped pyrophyllite increased and C 55 of K(I)- and Fe(II)-doped pyrophyllite decreased. The other elastic constants of the doped pyrophyllite demonstrated clear changes. The above results for all of the doped pyrophyllites indicated that the anti-deformation abilities of the a-axis and b-axis were stronger than that of the c-axis, and the deformation ability of the three axes of the undoped pyrophyllite was stronger than that of doped pyrophyllite. The shear deformation resistance of the (001) plane of the doped pyrophyllite was stronger than those of the (100) and (010) planes and weaker than that of the undoped pyrophyllite. The above results show that the effects of doping on the mechanical properties of pyrophyllite were significant.
The VRH approach (Hill, Reference Hill1952; Chung & Buessem, Reference Chung and Buessem1968) is a suitable method for calculating the B, G and Y of pyrophyllite. These parameters are crucial indicators of the mechanical behaviour of materials, and they play a significant role in characterizing their overall mechanical properties. The elastic parameters of the undoped and doped pyrophyllites are listed in Table 5. The B value of undoped pyrophyllite was 45.94 GPa, while the B values of Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllite were 40.93, 39.12, 44.66, 43.71 and 43.67 GPa, respectively. These results indicate that the resistance to volume change due to strain stress and fracture of doped pyrophyllite was weaker than that of undoped pyrophyllite. The G value denotes the tendency of an object to shear when the material is subjected to an opposing force. As listed in Table 6, the G values of all of the doped pyrophyllites were smaller than that of undoped pyrophyllite. In particular, the G value of K(I)-doped pyrophyllite decreased the most. The stiffness of a material can be illustrated by Young's modulus (Y). Compared with the Y value of the undoped pyrophyllite, the Y values of the Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllite decreased by 15.27%, 24.49%, 16.35%, 19.48% and 18.78%, respectively. These results demonstrate that the doping reduced the ability of pyrophyllite to resist external pressure and shear strain and reduced its rigidity, indicating that the pyrophyllite was easier to deform under external force after doping.
Table 5. The calculated elastic constants of the undoped and doped pyrophyllites.

Table 6. The calculated elastic mechanical parameters of the undoped and doped pyrophyllites.

The Poisson's ratio μ serves as an indicator of the bonding nature of materials (Yang et al., Reference Yang, Wang, Gan, Shi and Tang2018). The calculated μ value for the undoped pyrophyllite, as listed in Table 6, was found to be 0.20. This value suggests a strong covalent characteristic in the chemical bonding of pyrophyllite. Furthermore, as μ was less than the critical value of 0.27, this indicates that the material can be expected to be brittle. In terms of the effects of doping on pyrophyllite, the μ values of the Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllite increased to 0.22, 0.24, 0.24, 0.25 and 0.25, respectively. These results also indicate the strong covalent aspect to the chemical bonds of pyrophyllite after doping.
In addition, the empirical G/B ratio (Pugh, Reference Pugh2009; Fan et al., Reference Fan, Wei, Chai, Yu, Liu and Zhou2015) can serve as an indicator of the brittle or ductile behaviour of materials. When the G/B ratio is <0.57, this suggests a material will demonstrate ductile behaviour. Conversely, if the G/B ratio is >0.57, the material can be expected to exhibit brittle behaviour (Yang et al., Reference Yang, Wang, Gan, Shi and Tang2018). The G/B ratios of the undoped and doped pyrophyllites were greater than the critical value of 0.57, so they also showed brittle behaviour.
The vp and vs values are also listed in Table 6, and these are key quantities in the interpretation of seismic data. Compared with the undoped pyrophyllite, the obtained vp and vs values of the doped pyrophyllite were lower. In particular, the vp and vs values of K(I)-doped pyrophyllite decreased the most, demonstrating values of 5.04 and 2.96 km s–1, respectively. After doping, the Vickers hardness of pyrophyllite decreased, so the pyrophyllite gained a soapy and smooth surface, suggesting that doped pyrophyllite could be used in the pharmaceutical industry.
The degree of dependence of the modulus of elasticity of pyrophyllite in all directions can be determined from anisotropy data. Ranganathan & Ostoja-Starzewski (Reference Ranganathan and Ostoja-Starzewski2008) proposed an index of universal anisotropy that is applicable to all crystalline forms to assess differences in anisotropy, and this index is denoted by the symbol AU. The AU index is based on the upper and lower bounds of the bulk and shear moduli, and AU can be defined as per Equation 8:

The degree of deviation of the AU value from 0 reflects the degree of anisotropy of the material. To determine the anisotropy of pyrophyllite before and after doping, three-dimensional diagrams of the Young's modulus (Roman et al., Reference Roman, Pullumbi and Coudert2016) of undoped and Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllite were drawn, as shown in Fig. 4a–f. The three-dimensional diagrams of the Young's modulus were close to spherical, and the degree of irregularity of the sphere represented the degree of anisotropy of the material. In addition, the anisotropies of undoped and Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllite were calculated to be 8.04, 10.14, 8.31, 11.61, 14.95 and 12.72, respectively. These results indicate that both undoped and doped pyrophyllites showed anisotropy (Zhao & Xu, Reference Zhao and Xu2000), with the degree of anisotropy becomes greater after doping.

Figure 4. There-dimensional diagrams of the Young's modulus of the (a) undoped, (b) Na-doped, (c) K-doped, (d) Mg-doped, (e) Ca-doped and (f) Fe-doped pyrophyllites.
Conclusion
In this work, the effects of Na(I), K(I), Mg(II), Ca(II) and Fe(II) doping on the atomic structure, electronic properties and mechanical characteristics of pyrophyllite were determined using the DFT. The following conclusions can be drawn from the research:
(1) Compared to the atomic structure of the undoped pyrophyllite, the structures of the Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllites changed significantly. The influence of K(I) doping on the lattice parameters of pyrophyllite was the greatest, while the least influence was observed for Fe(II) doping. After doping, the orders of bond lengths of doping atoms with oxygen atoms were K–OH > Ca–OH > Na–OH > Mg–OH > Fe–OH > Al2–OH and K–Oa > Ca–Oa > Na–Oa > Mg–Oa > Fe–Oa > Al2–Oa. After doping, the layer thicknesses of Na(I)-, K(I)-, Mg(II)-, Ca(II)- and Fe(II)-doped pyrophyllites were reduced compared with that of the undoped pyrophyllite.
(2) In addition, the PDOSs, TDOSs and energy band structures of the five kinds of doped pyrophyllite also changed significantly. The order of impact on the band structure of pyrophyllite was Fe(II) > K(II) > Na(I) > Mg(I) > Ca(II). These features were caused by the differing electronegativities of the doping atoms and the oxygen atoms.
(3) Finally, the elastic constants of the doped pyrophyllites were lower than that of the undoped pyrophyllite. The deformation ability of the three axes of the doped pyrophyllites and the shear deformation resistance in the (001) plane of the doped pyrophyllites were weaker than those of the undoped pyrophyllite. Doping reduced the B, G, Y, vp and vs values but increased the AU value of pyrophyllite. The order of impact on the mechanical properties of pyrophyllite was Mg(II) > Fe(II) > Ca(II) > K(I) > Na(I).
The first-principles results obtained in the present work provide valuable insights in the physicochemical and mechanical properties of the substitutional defects in pyrophyllite at the molecular level.
Financial support
This work was supported by the National Natural Science Foundation of China (grant number 41702317) and Fundamental Research Funds for the Central Universities (2023ZKPYSB01).
Competing interests
The authors declare none.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations. Requests for data 6 months after publication of this article will be considered by the corresponding author.











