


INTRODUCTION
Campylobacter spp. are the foremost bacterial cause of gastroenteritis in the UK, with the estimated incidence being in the region of 300 000 cases per annum [Reference Adak1]. While poultry products are well recognized sources of human infection, there is increasing evidence that ruminants also play a role [Reference Karenlampi2, Reference Wilson3]. A recent study in Lancashire [Reference Wilson3] utilizing genotype data estimated that 35% [95% credible interval (CrI) 20·8–43·2] of human cases arose via acquisition of C. jejuni from cattle sources with 4·3% (95% CrI 0·1–17·5) from sheep sources. Poultry accounted for the majority of human cases (56·5% 95% CrI 51·1–61·8). Routes of ruminant-derived human infection include consumption of raw milk [Reference Gillespie4] and contamination of water sources [Reference Said5] although the precise infection routes remain to be elucidated in most cases. Studies from the UK [Reference Louis6] and New Zealand [Reference Gilpin7, Reference Mullner8] have suggested acquisition of infection via environmental exposures or contact with animals or their faeces may be important in rural settings, rather than just food sources.
Multi-locus sequence typing (MLST) [Reference Dingle9] based on the sequencing of fragments of seven housekeeping genes, is increasingly used in epidemiological studies of C. jejuni. Isolates are defined as sequence types (ST) by the allelic profiles of these seven genes. Sequence types may then be allotted to membership of a clonal complex (CC) defined as two or more independent isolates with sequence-type profiles that share identical alleles at four or more loci [of the seven loci sequenced].
Dingle et al. [Reference Dingle10] found six clonal complexes (CC ST21, CC ST45, CC ST206, CC ST61, CC ST48 and CC ST257) accounted for 60% of human disease isolates. A number of studies using MLST data have demonstrated that sequence types associated with human disease are widespread in the rural environment [Reference French11] and found in ruminants and wildlife [Reference Karenlampi2, Reference Dingle10, Reference Colles12, Reference Kwan13]. While there is considerable overlap between hosts, these studies suggest that sequence types belonging to the following clonal complexes predominate in cattle and sheep: CC ST61, CC ST42, CC ST21, CC ST48, while CC ST45 is predominantly isolated from chickens and wildlife but is also widespread in ruminants.
The objective of the present longitudinal study is to describe the spatio-temporal molecular epidemiology and population diversity of C. jejuni in cattle and sheep in a rural area of the UK. A previous analysis [Reference Grove-White14] of the complete dataset, in which the current dataset is nested, demonstrated peak prevalence of C. jejuni in both cattle and sheep occurring during the summer, although in cattle this apparent seasonality was associated with grazing pasture while in sheep it was independent of grazing. Increased prevalence was also associated with increased milk yield and herd size in dairy cattle, and with increased stocking density and pasture quality in sheep.
MATERIALS AND METHODS
The study design was a repeated cross-sectional study over a 2-year period starting in January 2006. Fifteen dairy and four sheep farms were recruited with the help of three spatially separated veterinary practices in Lancashire serving the Southern Fylde (zone 1), North Lancashire (zone 2) and Southeast Lancashire (zone 3) (Fig. 1). Eligible farms were dairy farms with >100 adult cows and sheep farms with >150 breeding ewes and no other livestock enterprises. Six dairy farms were recruited in zone 1 while four dairy and two sheep farms were recruited in each of the other zones. One farm in zone 2 ceased trading in December 2006 and was replaced with a neighbouring farm for the rest of the study. Another farm in zone 2 ceased keeping cattle in June 2007 thus sampling on this farm was incomplete. Farms were visited at 8-week intervals when 20 freshly voided faecal samples were collected from the lactating cows on dairy farms or adult sheep on sheep farms. Samples were only collected from animals observed to defecate by the author. Each faecal pat was sampled from at least three sites within the pat and mixed thoroughly in a sterile sample pot. Samples were transported to the laboratory on ice where 1 g faecal sample was placed in 9 ml Campylobacter enrichment broth (IDG Ltd, UK) with cefoperazone, vancomycin, trimethoprim and cycloheximide (CVTC supplement; IDG Ltd, UK), homogenized for 30 s in a Colworth 80 stomacher (A. J. Seward & Co. Ltd, UK) and then incubated for 24 h at 37°C in a variable atmosphere incubator (VAIN, Don Whitley Scientific Ltd, UK) under microaerobic conditions (12% CO2, 3% H2, 11% O2, 74% N2). Two Campylobacter blood-free selective agar (CSA) plates (IDG Ltd) enriched with cefoperazone and amphotericin (CA supplement; IDG Ltd) were inoculated with 50 μl and 5 μl, respectively of the enrichment broth and incubated under the same conditions for 60–72 h. Plates were then examined and up to four putative Campylobacter colonies (per faecal sample) were subcultured onto blood agar plates and incubated microaerobically for 72 h. Single colonies were then subcultured onto blood agar plates and incubated for a further 48 h as before.
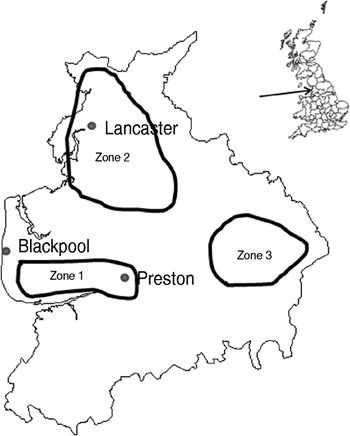
Fig. 1. Map of Lancashire showing approximate bounds of sampling zones.
A crude DNA aqueous lysate was prepared by inoculating 200 μl distilled water with a small amount of the culture, heating at 100°C for 15 min followed by centrifugation at 1300 rpm for 10 min. All putative Campylobacter isolates were frozen in Microbank tubes (Pro-Lab Diagnostics, UK) and stored at −80°C. PCR assays [Reference Gonzalez15–Reference Wang18] were employed for assignment to putative Campylobacter spp.
Of the 2307 C. jejuni isolates identified, 1003 isolates were selected for sequencing and subsequent assignment to sequence type. Isolates were randomly selected using Survey Toolbox [Reference Cameron19] stratifying by zone and sampling round (defined as the 8-week period during which each farm was sampled once).
Isolates were processed in batches of 250. DNA extraction was performed either by preparation of an aqueous lysate (first 500 isolates) or using a commercial kit (Prepman; Applied Biosystems, USA). The first 500 isolates were processed using primers and reaction conditions as described by Dingle et al. [Reference Dingle9] while the remaining isolates were processed using primers and reaction conditions according to Miller et al. [Reference Miller20]. The changes in protocol were adopted in an attempt to optimize the entire process. In the event of failure to obtain sequence data for a given allele, the entire process would be repeated for up to a maximum of six times. PCR amplification was performed in 50 μl volumes comprising 2 μl DNA lysate and 48 μl mastermix, using a programmable thermal cycler (ABI 20720; Applied Biosystems). Sequencing was performed using an ABI 3130xl automatic sequencer with a 50-cm array and POP-7 polymer (Applied Biosystems).
Sequence data assembly, alignment, and interrogation of the Campylobacter jejuni MLST website (http://pubmlst.org/campylobacter/) for assignment of sequence type was performed using the STARS computer program [Reference Man Suen and Ventress21]. Isolates were assigned to clonal complexes using BURST at http://pubmlst.org/. Data analysis was performed using Stata v. 10 (StataCorp, USA). The following information was collected at each sampling visit, via a short questionnaire delivered by the investigator: date of sampling, farm identity, geographical zone, species, number of animals in sampled group, daily average milk yield (dairy cows only), and sampling environment (at pasture or housed). In order to obtain unbiased prevalence estimates allowing for the stratified sampling scheme employed, stratum specific sampling probability weights, calculated as the inverse of the probability of a faecal pat being selected for MLST, taking host species into account, were employed in all analyses.
Since the C. jejuni isolates in this study were selected from the larger population collected during the study, we interpreted the term ‘prevalence’ as meaning the proportion of C. jejuni-positive pats which were colonized by a given clonal complex. Host species differences were investigated by univariable analysis, using robust standard errors, for clonal complexes with a prevalence of >3% in either host species, henceforth referred to as the ‘major clonal complexes’, namely CC ST21, CC ST42, CC ST45, CC ST48, CC ST52, CC ST61, CC ST206, CC ST257 and CC ST403.
Individual multivariable logistic regression models, with farm specified as a fixed effect, were fitted for each major clonal complex with the binary outcome variable being the presence or absence of the particular clonal complex in a faecal pat. Multivariable regression was not performed if the number of faecal pats containing the clonal complex of interest was <20, due to lack of statistical power, thus no modelling of sheep data was performed.
The following explanatory variables were included in the initial model: date of sampling, farm identity, geographical zone, species, number of animals in sampled group, daily average milk yield, sampling environment (at pasture or housed). A backward stepwise model-building strategy was employed to decide which variables to include in the final model, whereby a full model was built and then each variable removed in turn, a likelihood ratio test performed and the resultant P value noted. The variable with the highest P value was then omitted and the process repeated. This process was repeated until only variables with P<0·2 remained in the model. The omitted variables were then added back in turn, starting with the lowest P value, a likelihood ratio test performed after each addition, and the variable retained if P<0·2. This process was continued until no further variables could be added, to produce the final model. Interactions between variables in the final model were considered for inclusion and retained if they improved model fit. No significant interactions were identified. Model fit at each stage was assessed using the Bayesian Information Criterion (BIC) [Reference Long22]. For a model Mk with deviance D(Mk) the BIC is estimated as: BICk=D(Mk)−dfk * ln N, where dfk is the degrees of freedom associated with the deviance and N is the sample size. The more negative the BICk the better the model fit, with an absolute difference in BIC between two models of >6 offering strong support for the model with the smallest BIC.
Time was offered to the model as a composite of four sine and cosine functions (harmonic regression) to allow modelling of seasonal periodicity [Reference Stolwijk, Straatman and Zielhuis23]. Four time covariates (x 1, x 2, x 3, x 4) were generated as follows:
where t=week number, with week 1 being the first week in January 2006 when sampling commenced. This specification allows us to model seasonal effects more flexibly than would a simple harmonic (terms x 1 and x 2 only).
Gene flow was assessed by calculating Wright's F ST statistic [Reference Slakin and Barton24] both at sequence level using concentated sequence data for the seven alleles sequenced and at allelic profile level using the Arlequin v. 3.5 software package [Reference Schneider, Roessli and Excoffier25]. A value of 1 implies two populations are completely distinct with no flow of genes between them while a value of 0 implies the two populations are homogenous with similar allelic frequencies. Analysis of molecular variance (AMOVA) [Reference Excoffier, Smouse and Quattro26], a least squares methodology similar to ANOVA, was performed in Arlequin v. 3.5 using both allelic profiles and concentated sequence data for the seven alleles to investigate partitioning of the variance in molecular diversity.
Linkage disequilibrium was investigated by calculating the standardized Index of Association (IAS) [Reference Maynard Smith27] in the software package LIAN [Reference Haubold and Hudson28].
Diversity at sequence level was described by calculating Simpson's diversity index (DI) [Reference Simpson29]. It is calculated as follows:
where N=total number of isolates and n=number of isolates belonging to a particular sequence type.
DI may take a value between 0 and 1, where 0 represents no diversity and 1 represents infinite diversity. Analytical rarefaction to produce rarefaction curves [Reference Hughes30] was performed to further investigate sequence-type diversity. The proportional similarity index (PSI) or Czekanowski index [Reference Rosef31] was also calculated. This is an objective and simple measure of the area of intersection of two frequency distributions and provides an estimate of the similarity of sequence types from different sources. It is calculated by: PSI=1−0·5 Σi|p i−q i|, where p i and q i are the proportions of strains belonging to type i out of all strains typed from sources P and Q. The value for PSI ranges from 1 for identical frequency distributions to 0 for distributions with no common types.
RESULTS
Twenty faecal samples were collected at each sampling visit, yielding a total of 4260 samples. Four potential isolates were taken from each sample equating to 17 040 potential bacterial isolates. In total, 9499 putative Campylobacter spp. isolates were grown and 2307 (24·3%) were identified as C. jejuni. At pat level, this equated to an estimated C. jejuni faecal pat prevalence of 19·1% (95% CI 15·4–22·7) and 17·0% (95% CI 8·5–25·5) for cattle and sheep, respectively.
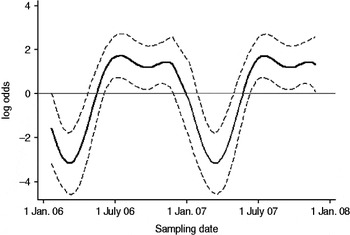
Fig. 2. The seasonal component to variation in C. jejuni MLST CC ST45 faecal pat prevalence on Lancashire dairy farms. The estimated seasonal effect is shown as a solid line, with dashed lines indicating pointwise upper and lower 95% confidence limits.
Of the 2307 C. jejuni isolates identified 1003 were sequenced. Full allelic profiles were obtained for 939 isolates while partial profiles were obtained for 64 isolates. A total of 154 sequence types were identified with 86 being new sequence types. Of the 1003 C. jejuni isolates sequenced, 856 were assigned to existing clonal complexes (C. jejuni MLST database interrogated on 19 June 2008) (Supplementary Fig. 1, available online). Seven hundred and eighty-five bovine isolates with full allelic profiles were obtained from 485 faecal pats while 154 ovine isolates were obtained from 106 pats, making an overall total of 591 colonized pats. One hundred and thirty-four (22·7%) pats were colonized by more than one species of Campylobacter. Three hundred and six pats (52%) yielded one isolate of C. jejuni with 181 yielding two isolates, 81 yielded three isolates while 23 pats yielded four isolates. Four hundred and eighty-nine pats contained one sequence type only while 65 contained two sequence types, six pats contained three sequence types and two pats contained four sequence types. Therefore in total, 73 pats contained multiple sequence types (Table 1).
Table 1. Pat-level distribution of sequence types in faecal pats from cattle and sheep faecal pats collected in Lancashire
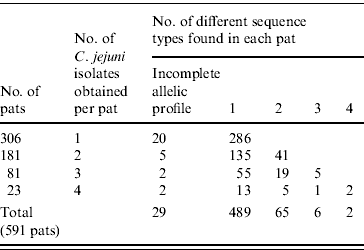
Of the 73 pats containing more than one sequence type, 37 (51%) contained sequence types belonging to the same clonal complex with 32 pats (44%) containing sequence types belonging to different clonal complexes while four pats contained sequence types unassigned to clonal complexes.
Eighty-seven percent of all isolates sequenced belonged to the following clonal complexes: CC ST21, CC ST42, CC ST45, CC ST48, CC ST52, CC ST61, CC ST206, CC ST257 and CC ST403. There was considerable between-farm variation both in presence and prevalence of individual clonal complexes with CC ST61 being the only clonal complex isolated from all farms (Supplementary Figs 2, 3, online).
In cattle faeces, CC ST61 (25·5%) was the predominant clonal complex, followed by CC ST21 (19·8%) and CC ST403 (15·9%). In sheep faeces, the predominant clonal complexes, namely CC ST21, CC ST42, CC ST48, CC ST52 were isolated with frequencies ranging from 14% to 18% (Table 2). Univariate analysis, taking sampling strategy into account demonstrated apparent host species differences in clonal complex faecal pat prevalences with CC ST403 being higher in cattle faeces (P=0·008) while CC ST48 and CC ST52 were found at higher prevalences in sheep faeces (P=0·007 and P=0·001, respectively) with CC ST257 only being found in cattle faeces (Table 3). In the case of cattle isolates, multivariable logistic regression with farm specified as a fixed effect was performed to investigate any associations between farm identity, herd size, milk yield, geographical zone, date of sampling, sampling environment and presence of a given clonal complex. For all clonal complexes investigated apart from CC ST45, farm identity was the only significant covariate associated with faecal pat clonal complex prevalence. In the case of CC ST45, prevalence was also strongly associated with time of sampling, with peak prevalence occurring during the summer months (Fig. 2).
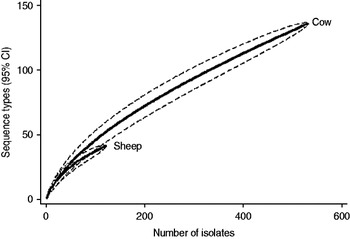
Fig. 3. Rarefaction curves for all isolates by host species. Dashed lines indicate the upper and lower 95% confidence limits.
Table 2. Pat-level distribution of C. jejuni MLST clonal complexes in cattle and sheep faecal pats from Lancashire
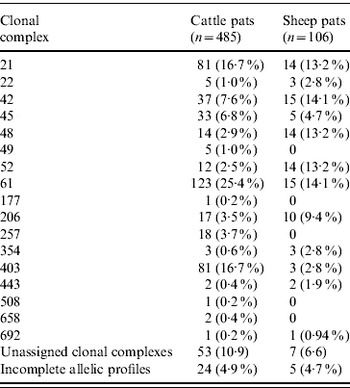
Table 3. Estimated faecal pat prevalenceFootnote * of the major C. jejuni MLST clonal complexes in cattle and sheep faecal pats from Lancashire
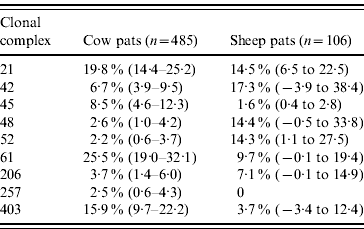
Values in parentheses are 95% confidence intervals.
* Prevalence: as estimated after taking into account the stratified sampling strategy employed.
Gene flow was assessed at sequence and allelic profile level by calculating the pairwise Wright's F ST statistic as follows: between farms, between geographical zones, between host species and between animals grazing outside and animals kept inside. There was little difference between F ST values estimated at sequence level compared to allelic profile level. Pairwise F ST values between farms varied from 0·01 to 0·50 demonstrating the presence of heterogeneity at farm level, suggesting some population differentiation (Supplementary Fig. 4, online). At between-group level, F ST values ranged from 0·16 to 0·19 suggesting little heterogeneity is present at these levels.
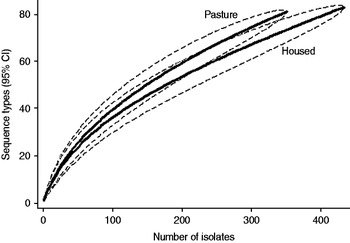
Fig. 4. Rarefaction curves for cattle isolates by sampled environment. Dashed lines indicate the upper and lower 95% confidence limits.
AMOVA was performed at both sequence and allelic profile level, to investigate partitioning of molecular variance at four levels, namely between zone, between farm, between animal species, within farm. In all analyses performed most variation (80–84%) was at the within-farm level (P<0·001) with 16–20% of variation at the between-farm level (P<0·001) with minimal variation (<2%) residing at the higher group level (between geographical zones, between host species and between animals grazing outside and animals kept inside).
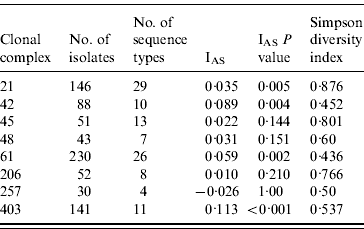
The standardized IAS was significantly (P<0·001) greater than zero for the entire dataset (0·498) demonstrating the presence of linkage disequilibrium. Similarly, linkage disequilibrium within a clonal complex was demonstrated for CC ST21, CC ST42, CC ST61 and CC ST403 but not for CC ST45, CC ST48, CC ST206 or CC ST257 (Table 4). In all cases, the clonal complex level IAS values (−0·03 to 0·11) are considerably less than the IAS for the entire dataset.
Table 4. Genotypic diversity in C. jejuni isolates from cattle and sheep in Lancashire
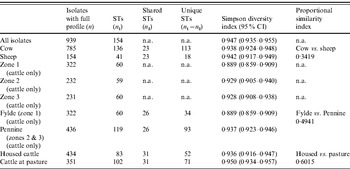
ST, Sequence type; CI, confidence interval; n.a., not available.
Simpson's DI (Table 5) for the entire dataset was 0·947 (95% CI 0·935–0·955). There was considerable between-farm variation in diversity as demonstrated by the wide range of DI estimates obtained (0·65–0·916).
Table 5. Indices of Association (IAS) and Simpson diversity indices for C. jejuni isolates belonging to the major clonal complexes, isolated from cattle and sheep in Lancashire
Examination of the relevant rarefaction curves suggest greater sequence-type diversity in bovine isolates compared to sheep isolates (Fig. 3); in bovine isolates collected at pasture compared to those collected from housed animals (Fig. 4); and in bovine isolates collected in zone 2 (North Lancashire) and zone 3 (Southeast Lancashire) compared to those collected in zone 1 (South Fylde) (Fig. 5). The rarefaction curves for zones 2 and 3 were almost identical suggesting similar levels of sequence-type diversity in these areas, both of which lie in the foothills of the Pennines.
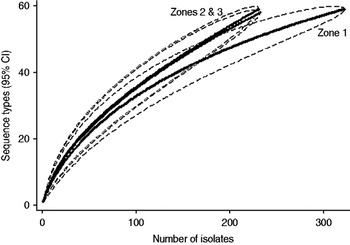
Fig. 5. Rarefaction curves for cattle isolates by geographical zone. Dashed lines indicate the upper and lower 95% confidence limits. Zone 1, Fylde; zone 2, North Lancashire; zone 3, Southeast Lancashire.
The PSI was 0·35 between cattle and sheep isolates suggesting a moderate degree of overlap between the two host species. There was considerable overlap between bovine isolates from animals at pasture compared to housed animals (PSI=0·60), and between bovine isolates collected in the Southern Fylde (zone 1) and those collected from the foothills of the Pennines (zones 2 and 3) (PSI=0·49).
Diversity was greatest in isolates belonging to CC ST21 and CC ST45 compared to the other major clonal complexes (Table 4).
DISCUSSION
This study represents the largest MLST-based study of C. jejuni in cattle and sheep at the present time, with 939 isolates assigned to sequence type, representing 41% of the total C. jejuni isolates collected over a 2-year period from 15 dairy and four sheep farms. The 939 isolates were assigned to 68 existing and 86 new sequence types suggesting that there is considerably more diversity in cattle and sheep C. jejuni isolates than is currently recorded on the pubmlst database (http://pubmlst.org/ campylobacter/).
Eighty-seven percent of the isolates in the present study belonged to CC ST21, CC ST42, CC ST45, CC ST48, CC ST52, CC ST61, CC ST206, CC ST257 and CC ST403 which is in broad agreement with previous studies [Reference Karenlampi2, Reference Dingle10–Reference Kwan13]. All of these clonal complexes have been found in association with human disease [Reference Dingle10, Reference Sopwith32]. In contrast to previous studies [Reference Dingle10, Reference French11], which have suggested that CC ST45 is associated primarily with poultry, wildlife, water and human cases, the present study found it to be well represented in bovine faecal isolates.
A marked finding in the present study was the considerable between-farm variation in terms of both the presence of a clonal complex and its prevalence, e.g. CC ST61 was the only clonal complex to be isolated from all farms, while CC ST257 was only isolated from six of the 15 dairy farms and none of the sheep farms. Similar findings have been observed in a 12-month longitudinal study of five dairy farms in the Wirral, Cheshire [Reference Kwan13].
Multivariable analysis demonstrated that in the case of CC ST21, CC ST42 and CC ST61, the variation in prevalence observed was associated primarily with farm identity alone, i.e. localized spatial variation while in the case of CC ST45, in addition to localized spatial variation, there was also a significant temporal association with a higher prevalence observed during the summer months. A study of 493 human C. jejuni isolates collected during 2003–2004 from Lancashire [Reference Sopwith32] found CC ST21 followed by CC ST45 and CC ST257 to be the chief MLST clonal complexes causing notified cases of human disease. Cases due to CC ST45 were most often reported from rural areas during the summer months, in particular during June and July, corresponding to the period of increased prevalence of CC ST45 observed in cattle in the present study. The observation that CC ST45 has been shown to be the predominant C. jejuni clonal complex to be isolated from recreational surface water in the same study area [Reference Sopwith33], suggests a possible transmission pathway between bovine faeces and human illness due to CC ST45 in the summer months. It is estimated that 100 cows will produce upwards of 5000 l of faeces daily [34] thus offering considerable potential for contamination of surface water sources.
There were host species differences in prevalence, with CC ST45, CC ST257 and CC ST403 being more frequent in cattle isolates, while CC ST48 and CC ST52 were more widespread in the sheep isolates. However, there was no difference in the frequency of isolation of CC ST21, CC ST42, CC ST61 or CC ST206 between cattle and sheep.
CC ST61 was the most frequently isolated complex overall, confirming its pre-eminence in ruminants and adding weight to the hypothesis that it is a ruminant-adapted strain [Reference French11, Reference Colles12].
The findings regarding the host distribution of CC ST21 support the observations of French et al. [Reference French11] who found it to have no apparent host preference, being isolated at similar frequencies from cattle and wildlife samples in a cross-sectional study of a 100 km2 area of farmland in Cheshire. A similar conclusion was drawn by Colles et al. [Reference Colles12] who speculated that members of CC ST21 may be well adapted for long-term survival given their apparently ubiquitous distribution.
Sampling strategy is crucial in recognition and quantification of microbial diversity at both within-animal and between-animal levels. For within-animal diversity this implies taking multiple isolates from the animal or in this case of the current study, from the faecal pat. In the present study only four putative Campylobacter colonies were selected from each faecal pat. This decision was made primarily on logistical grounds. Despite the limitations of our sampling strategy we identified colonization by more than one sequence type in 73 (12%) of the 591 sampled pats. Approximately half were colonized by sequence types belonging to the same clonal complex with most of the remainder being colonized by sequence types belonging to different clonal complexes. This would suggest that there is both substantial sequence type and clonal complex diversity present within animals as well as between animals. Further studies are required to quantify this diversity. The issue of optimal microbial sampling strategy has been addressed previously [Reference Singer35, Reference Dopfer36].
The standardized IAS for the entire dataset was significantly different from zero, which is consistent with the presence of linkage disequilibrium and a weakly clonal population structure as described previously [Reference Dingle10]. At the individual major clonal complex level the IAS values were smaller by an order of magnitude than for the entire dataset. The IAS values for CC ST45, CC ST48, CC ST206 and CC ST257 were not significantly different to zero suggesting these populations are in linkage equilibrium implying higher levels of recombination, whereas IAS values for CC ST21, CC ST42, CC ST61 and CC ST403 were significantly greater than zero implying lower rates of recombination and the presence of linkage disequilibrium. Similar findings have been reported for CC ST21 and CC ST45 in poultry [Reference Habib37].
There was considerable between-farm variation in sequence-type diversity, as demonstrated by the range of individual farm Simpson indices (0·65–0·916). Sequence-type diversity was greater in cattle isolates compared to sheep isolates and there was significantly greater diversity in bovine isolates collected at pasture compared to when housed. In a previous analysis [Reference Grove-White14] of the entire dataset from which the current dataset is derived, we reported that the bovine faecal pat prevalence of C. jejuni was significantly higher at pasture. Thus, both prevalence and diversity appeared to be greater in cattle at pasture compared to indoors. Two hypotheses may be generated to explain this apparent association. First, the increased diversity observed is due to the increased prevalence of carriage of the organism observed at pasture resulting in greater opportunity for recombination. Second, an alternative hypothesis is that there is increased transmission from wildlife and birds to cattle when they are outside compared to when housed. The observation of increased diversity in isolates belonging to CC21 and CC45 compared to other clonal complexes is of particular interest in this respect since both clonal complexes are widespread in wildlife and birds.
There appeared to be greater sequence-type diversity in cattle isolates present within zones 2 and 3 compared to zone 1. It may be hypothesized that this is a reflection of possibly greater biodiversity and increased wildlife in the foothills and valleys of the Pennines compared to the Southern Fylde, a low-lying, intensively farmed, flat grassland and vegetable-growing area with relatively little woodland. It is recognized that intensive farming and monoculture are associated with reduced biodiversity [Reference Krebs38] and latterly EU and UK government policy in areas such as the Pennines has actively supported farming practices to encourage biodiversity via various financial grants. Increased biodiversity might result in higher rates of transmission of campylobacters between cattle, sheep and wildlife.
Both gene flow and AMOVA demonstrate that the majority of genetic variation (~80%), whether at sequence or allelic profile level, resides at the within-farm level but there is a degree of heterogeneity with population differentiation at the between-farm level. This supports the findings reported at clonal complex level. However, there was no evidence of population differentiation at higher levels such as species, geographical zone or sampled environment.
Recombination and generation of sequence-type diversity in C. jejuni in farm animals will be favoured by increased population sizes favouring between-animal transmission and co-colonization by multiple strains. Herd and flock size have increased dramatically during this period particularly since 1945 when UK agriculture was encouraged to expand and intensify. This trend towards increased dairy herd size accelerated considerably in the 1980s and continues today and is likely to have driven sequence-type diversity.
This study has highlighted the genetic diversity in C. jejuni isolates from cattle and sheep and, furthermore, has demonstrated the considerable variation in diversity at farm level. This observed diversity is likely to be a reflection of both past and present events in terms of recombination, mutation, transmission barriers, bottlenecks and influx of strains from external sources such as wildlife. A recent study [Reference Wilson39] of human C. jejuni isolates from the same geographical area as the present study lends support to the hypothesis that diversity, as detected by MLST, is generated more rapidly than previously thought. That study estimated the time of importation of several C. coli alleles including uncA-17 [Reference Dingle40] into ST61 (the most prevalent sequence type in our study) to have occurred just decades ago, suggesting that the rate of recombination in C. jejuni is such that evolutionary change can be detected in real time. Our study has demonstrated that the majority of genetic diversity in C. jejuni populations resides at the within-farm level. Greater understanding of farm-level effects in driving this diversity is paramount for control of zoonotic organisms such as Campylobacter spp. in others.
NOTE
Supplementary material accompanies this paper on the Journal's website (http://journals.cambridge.org/hyg).
ACKNOWLEDGEMENTS
This work is dedicated to the memory of the late Professor Tony Hart who died suddenly in 2007. Before his death he was intimately involved with the work presented here. This study was funded by Defra and HEFCE as part of the Veterinary Training and Research Initiative (VTRI). We thank Ms. J. Sutherst, Ms. P. Houghton and Ms. A. M. Riley for laboratory assistance. We also thank the veterinary surgeons and farmers who participated in the study.
DECLARATION OF INTEREST
None.













