


Introduction
The mitochondrial genome (mitogenome) encodes proteins that are involved in electron transport chain and oxidative phosphorylation that are essential for the energy production of the cell (Wolstenholme, Reference Wolstenholme1992; Cameron, Reference Cameron2014). Insect mitogenomes are circular double-stranded DNA (dsDNA) molecules that typically range in size from 14 to 20 kb (Boyce et al., Reference Boyce, Zwick and Aquadro1989; Wolstenholme, Reference Wolstenholme1992) and contain a conserved set of 13 protein coding genes (PCGs), 22 transfer RNAs (tRNAs), and 2 ribosomal RNA genes (rRNAs) that are required for the translation of mitogenome-encoded proteins (Boore, Reference Boore1999). Insect mitogenomes usually contain a large A+T-rich, non-coding control region that functions in the initiation of transcription and the replication of the mtDNA (Wolstenholme, Reference Wolstenholme1992; Cameron, Reference Cameron2014), where the length is highly variable among different lineages, due to high rates of nucleotide substitution, insertions/deletions, and variable numbers of tandem repeats (Fauron and Wolstenholme, Reference Fauron and Wolstenholme1980; Inohira et al., Reference Inohira, Hara and Matsuura1997). Animal mitogenomes have a highly conserved gene content, maternal inheritance, and lack recombination (Avise, Reference Avise2000), which have facilitated their use in the studies of phylogenetic relationships, population genetic structuring, as well as comparative evolution (Zhang et al., Reference Zhang, Lu, Zhao, Zhang, Diasio, Habus, Jiang, Iyer, Yu and Agrawal1995; Nardi et al., Reference Nardi, Carapelli, Dallai and Frati2003; Arunkumar et al., Reference Arunkumar, Metta and Nagaraju2006). Comparisons between various insect mitogenomes have enabled the estimation of short-term evolutionary patterns within and between closely related species (Ballard, Reference Ballard2000; Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005). Although the gene content of the metazoan mitogenomes is highly conserved, variable lengths of A+T-rich coding regions, gene order, and orientation of tRNAs and PCGs are observed among insects (Hong et al., Reference Hong, Jiang, Yu, Yang, Li, Xue and Wei2009).
To date, complete or near complete mitogenomes have been sequenced from more than 600 arthropod species (Cameron, Reference Cameron2014). Although the insect order Lepidoptera contains approximately 200,000 species, 72 complete or near complete mitogenomes representing 17 families in the lepidopteran lineage Ditrysia are available (Cameron, Reference Cameron2014). Among the family Crambidae, mitogenomes have been obtained for eight species: Ostrinia nubilalis and O. furnacalis (Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005), Diatraea saccharalis (Li et al., Reference Li, Gao, Liu, Liu, Liang, Zhou and Cai2011), M. vitrata (Old World) (Margam et al., Reference Margam, Coates, Hellmich, Agunbiade, Seufferheld, Sun, Ba, Sanon, Binso-Dabire, Baoua, Ishiyaku, Covas, Srinivasan, Armstrong, Murdock and Pittendrigh2011b ), Cnaphalocrocis medinalis (Chai et al., Reference Chai, Du and Zhai2012), Chilo suppressalis (Chai et al., Reference Chai, Du and Zhai2012), Paracymoriza prodigalis (Ye et al., Reference Ye, Shi, Xing, Yu and You2013), and Spoladea recurvalis (He et al., Reference He, Zou, Zhang, Ma, Zhang and Yue2015). Complete mitogenome sequences for Lepidoptera have been used to study the divergences between sibling and congeneric species (Yukuhiro et al., Reference Yukuhiro, Sezutsu, Itoh, Shimizu and Banno2002; Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005) and to facilitate population-level studies (Kim et al., Reference Kim, Lee, Seol, Yun, Lee, Hwang and Jin2006). Furthermore, full mitogenomes for Lepidoptera are important for the determination of phylogenetic relationships and the identification of cryptic species (Lee et al., Reference Lee, Shin, Kim, Park, Cho and Kim2006; Chai et al., Reference Chai, Du and Zhai2012) at various taxonomic levels.
Obtaining full mitogenome sequences has been a challenging and resource demanding task when sequenced by long-range PCR and subsequent primer walking (Huyse et al., Reference Huyse, Plaisance, Webster, Mo, Bakke, Bachmann and Littlewood2007) or overlapping short polymerase chain reaction (PCR) products (Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005). Recently, next-generation sequencing (NGS) technologies have led to more straightforward pipelines for the assembly of complete mitogenome sequences (Jex et al., Reference Jex, Hall, Littlewood and Gasser2010a ; Knaus et al., Reference Knaus, Cron, Liston, Pilgrim and Schwartz2011). Mitogenomes have been sequenced from NGS libraries prepared from long-PCR amplified sequences (Maricic et al., Reference Maricic, Whitten and Pääbo2010; Morin et al., Reference Morin, Archer, Foote, Vilstrup, Allen, Wade, Durban, Parsons, Pitman, Li, Bouffard, Abel Nielsen, Rasmussen, Willerslev, Gilbert and Harkins2010; Horn et al., Reference Horn, Durka, Wolf, Ermala, Stubbe, Stubbe and Hofreiter2011) or from sequence captured DNA molecules (Vasta et al., Reference Vasta, Ng, Turner, Shendure and Hahn2009). Long-range PCR strategies may remain useful for NGS-based sequencing, especially when limited genetic material can be recovered from small invertebrates (Jex et al., Reference Jex, Littlewood and Gasser2010b ). Ultra-deep sequencing on NGS platforms can acquire data that are sufficient for the subsequent assembly of full mitogenomes at high read depth (Knaus et al., Reference Knaus, Cron, Liston, Pilgrim and Schwartz2011). Also, due to the high copy numbers of mitochondria in most eukaryotic cells, NGS can reduce the impact of rare nuclear-integrated copies of mitochondrial DNAs or other non-target products (Gilbert et al., Reference Gilbert, Tomsho, Rendulic, Packard, Drautz, Sher, Tikhonov, Dalén, Kuznetsova, Kosintsev, Campos, Higham, Collins, Wilson, Shidlovskiy, Buigues, Ericson, Germonpré, Götherström, Iacumin, Nikolaev, Nowak-Kemp, Willerslev, Knight, Irzyk, Perbost, Fredrikson, Harkins, Sheridan, Miller and Schuster2007; Ho and Gilbert, Reference Ho and Gilbert2010).
Members from the genus Maruca (Lepidoptera: Crambidae) are serious insect pests that feed on cowpea crops throughout the tropical and subtropical regions, from northern Australia and East Asia through sub-Saharan Africa to the Caribbean, Central America and Hawaii (Sharma, Reference Sharma1998). Feeding by M. vitrata causes considerable crop loss and reduced yields to cultivated cowpea in many African countries, and is a target for improved and sustainable control methods (Agunbiade et al., Reference Agunbiade, Steele, Coates, Gassmann, Margam, Ba, Dabire-Binso, Baoua, Bello-Bravo, Seufferheld, Sun, Tamò, Pittendrigh, Boukar, Coulibaly, Fatokun, Lopez and Tamò2012). The comparison of M. vitrata mitochondrial cytochrome c oxidase subunit I (cox1) sequence data obtained from samples collected in Africa and Puerto Rico revealed unexpectedly high levels of sequence variation, which suggested the existence of cryptic species or undefined strains (Margam et al., Reference Margam, Coates, Ba, Sun, Binso-Dabire, Baoua, Ishiyaku, Shukle, Hellmich, Covas, Ramasamy, Armstrong, Pittendrigh and Murdock2011a ). A recent study also confirmed the existence of different subspecies of M. vitrata in Asia and Africa and Maruca spp. in Oceania and Latin America based on cox1 data (Malini et al., Reference Malini, Schafleitner, Muthukalingan and Ramasamy2015). An analogous relationship among New and Old World species has also been predicted between Helicoverpa zea and H. armigera, the corn earworm, and cotton bollworm (Behere et al., Reference Behere, Tay, Russell, Heckel, Appleton, Kranthi and Batterham2007), but accurate estimates of time since divergence between these species are lacking. As a result of the lack of morphological characters for subspecies sorting between the Old and New World strains of M. vitrata, Illumina HiSeq 2000 sequencing reads were obtained from M. vitrata (New World) and used to successfully assemble the full mitogenome sequence. These data were applied to refine previous haplotype divergence estimates and provide an estimated time of divergence between the New and Old World strains, in ongoing efforts to describe the evolutionary divergence in M. vitrata.
Materials and methods
Sample collection and DNA extraction
Insect samples of M. vitrata adults were collected from white bean (Phaseolus vulgaris) near Lares, Puerto Rico. All specimens were preserved in 100% ethanol and stored at 4 °C. Total genomic DNA was extracted from the adult specimens using the DNeasy animal tissue kit following the manufacturer's instructions (QIAGEN, Valencia, California). The quality of the DNA was assessed through electrophoresis on a 1% agarose gel before submission for library construction and sequencing.
Illumina HiSeq 2000™ library construction and sequencing
A shotgun genomic DNA library was constructed using the TruSeq DNA sample prep kit (Illumina, San Diego, California). Briefly, 1 μg of genomic DNA was sonicated on a Covaris M220 (Covaris, Woburn, Massachusetts) using the default methods for sonication to a size of 500 bp library, as described by the manufacturer. Sonicated DNA was blunt-ended, 3′-end A-tailed and ligated to an indexed adaptor. The adaptor-ligated gDNA was amplified by PCR to selectively enrich for those fragments that have adapters on both ends. Amplification was carried out for eight cycles with the Kapa HiFi polymerase (Kapa Biosystems, Woburn, Massachusetts), to reduce the likeliness of multiple identical reads, due to preferential clonal amplification. The library was size selected on a 2% agarose gel for fragments 600 bp to 800 bp in length. The size-selected library was run on Agilent bioanalyzer DNA 7500 LabChips (Agilent, Santa Clara, California), to determine the average fragment size and to confirm the presence of DNA of the expected size range, and quantitated by qPCR on an ABI 7900. The indexed library was loaded onto a single lane of an eight-lane flowcell for cluster formation and paired-end sequenced at 100-bp lengths on an Illumina HiSeq 2000™ using the TruSeq SBS sequencing kits version 3. The raw .bcl files were converted into FASTQ files using the software Casava 1.8.2 (Illumina). All shotgun genomic DNA library construction and Illumina HiSeq 2000™ sequencing were carried out at the W. M. Keck Center for Comparative and Functional Genomics, Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign.
Mitogenome assembly
FASTQ output was trimmed of read data with PHRED quality scores < 20 using the script TrimmingReads.pl (Patel and Jain, Reference Patel and Jain2012; NGSToolKit). A total of 30 million reads from the trimmed FASTQ sequence were loaded into the Velvet Assembler program v.1.2.10 (Zerbino and Birney, Reference Zerbino and Birney2008) and assembled using the de Bruijn graph method with a hash size (k-mer) = 21, no coverage cutoff, and a minimum contig length (−min_contig_lgth) = 10,000. Querying the National Center for Biotechnology Information (NCBI) non-redundant (nr) nucleotide database with the blastn algorithm (Altschul et al., Reference Altschul, Madden, Schäffer, Zhang, Zhang, Miller and Lipman1997; http://blast.ncbi.nlm.nih.gov/Blast.cgi) identified resulting contigs containing mitogenome sequence. The contigs that produced database ‘hits’ were filtered for those with E-values ≥ 10−40 and percent similarities ≥ 75 to identify contigs with encoded mitochondrial gene sequences.
Sequence annotation
Coding sequences for each M. vitrata (New World) mitochondrial gene were aligned to mitochondrial gene sequences from crambid mitogenomes (GenBank Accessions: JF339041, JN246082, FJ240227, HM751150, AF467260, NC_020094, and KJ739310) (Supplementary Table 1) using the CLC Genomics Workbench v6.5.2. The 5′ end of the mitochondrial genes was inferred to be the first legitimate start codon in the open reading frame, and manual adjustments were made when needed. The 3′ end was inferred to be at the first in-frame stop codon encountered. When the stop codon was located within the sequence of a downstream gene encoded on the same strand, a truncated stop codon (T) adjacent to the beginning of the downstream gene was designated as the termination codon. The positions and secondary structures for the tRNA genes were confirmed using ARWEN v1. 2 (Laslett and Canbäck, Reference Laslett and Canbäck2008; http://mbio-serv2.mbioekol.lu.se/ARWEN/index.html). The graphical circular structure of the mitogenome was drawn using GenomeVx, a suite of tools for generating physical maps of plastid and mitogenomes (Conant and Wolfe, Reference Conant and Wolfe2008; http://wolfe.ucd.ie/GenomeVx/). All annotations were transferred to Sequin 13.05 (http://www.ncbi.nlm.nih.gov/Sequin/) and submitted to the GenBank database under the accession number KJ466365.
Genome sequence analysis
The nucleotide sequences, codon usage, and relative synonymous codon usage (RSCU) in the M. vitrata (New World) mitogenome were calculated with MEGA 5.2.2 (Tamura et al., Reference Tamura, Nei and Kumar2004). Nucleotide composition skew analysis was carried out by the formula: AT skew = [A−T]/[A+T] and GC skew = [G−C]/[G+C], respectively (Perna and Kocher, Reference Perna and Kocher1995).
The DNA and protein sequence identities were calculated across the Crambidae using the FASTA program, a tool for biological sequence comparison (Pearson and Lipman, Reference Pearson and Lipman1988; http://fasta.bioch.virginia.edu/fasta_www2/). The online program, microsatellite repeats finder (http://insilico.ehu.es/mini_tools/microsatellites/) was used to analyse the A+T-rich control region for tandem repeats (repeat units ≥ 2 nucleotides and arrays of 4 ≥ units).
Estimation of divergence times
The nucleotide sequence of M. vitrata (Old World; GenBank accession number HM751150) was downloaded in the FASTA format and aligned with that of the M. vitrata New World sample (GenBank accession number KJ466365) using MEGA 5.2.2 (Tamura et al., Reference Tamura, Nei and Kumar2004). Gaps in the resulting alignment due to ambiguous bases (Ns) in HM751150 overhangs were deleted. The number of nucleotide differences between the two aligned sequences were calculated using DiffSeq from the EMBOSS Package (Rice et al., Reference Rice, Longden and Bleasby2000). A clock-like rate of 2% mitochondrial DNA sequence change per million years has been estimated among Drosophila species (Powell et al., Reference Powell, Caccone, Amato and Yoon1986) and a 1.115% per million year estimate was proposed among Lepidoptera (Brower, Reference Brower1994), which to date is the divergence time based on the substitution rate across a 11,250 bp of the consensus alignment between M. vitrata strains (inter-species substitution rate/clock rate).
Results and Discussion
Mitogenome assembly and annotation
Paired-end Illumina sequencing generated a total of 336,472,660 reads, of which 30 million were subsampled and used to generate a 15,385 bp de novo assembly of the previously uncharacterized M. vitrata (New World) mitogenome (Table 1 and Fig. 1). The mitogenome of M. vitrata (New World) was similar to the size previously reported from other Lepidoptera, that range from 15,314 bp in Coreana raphaelis (Lepidoptera: Lycaenidae) (Kim et al., Reference Kim, Lee, Seol, Yun, Lee, Hwang and Jin2006) to 15,928 bp in Bombyx mandarina (Lepidoptera: Bombycidae) (Yukuhiro et al., Reference Yukuhiro, Sezutsu, Itoh, Shimizu and Banno2002). The M. vitrata (New World) mitogenome contained the 13 PCGs, 22 tRNAs, and 2 rRNA genes that are typical for bilaterian metazoans (Wolstenholme, Reference Wolstenholme1992; Table 1 and Fig. 1). As in all other lepidopteran mitogenomes, of the total 37 genes, 23 were encoded on the major strand, whereas the 14 others were encoded on the minor strand (Table 1 and Fig. 1). The 13 PCGs spanned 11,369 bp and accounted for 73.90% of the entire sequence. The M. vitrata (New World) mitogenome had the typical Ditrysian gene order trnM-trnI-trnQ-nad2 (Yukuhiro et al., Reference Yukuhiro, Sezutsu, Itoh, Shimizu and Banno2002; Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005; Kim et al., Reference Kim, Lee, Seol, Yun, Lee, Hwang and Jin2006; Lee et al., Reference Lee, Shin, Kim, Park, Cho and Kim2006; Cameron and Whiting, Reference Cameron and Whiting2008; Hong et al., Reference Hong, Lee, Jo, Park, Kim, Hwang, Jin, Kang, Kim, Han and Kim2008), which differed from the ancestral gene order trnI-trnE-trnM-nad2 found in other insects (Cameron, Reference Cameron2014). This was first reported by Taylor et al. (Reference Taylor, McKechnie, Pierce and Kreitman1993) in lycaenids and noctuids, but has now been found in five lepidopteran superfamilies (Papilionoidea, Noctuoidea, Pyraloidea, Tortricoidea, and Bombycoidea) and a synapomorphy of Ditrysia (Salvato et al., Reference Salvato, Simonato, Battisti and Negrisolo2008).
Table 1. Annotation of the full mitogenome of Maruca vitrata (New World)
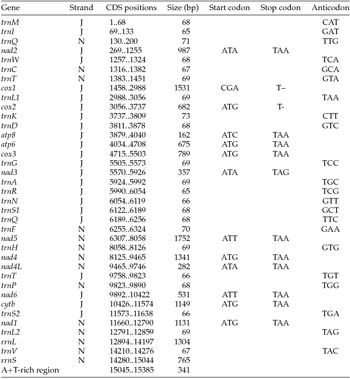
J, major strand; N, minor strand.
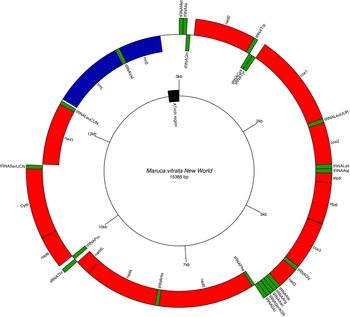
Fig. 1. Gene arrangements in the circular mitogenome of Maruca vitrata (New World). Red colour indicates the protein coding genes; green, tRNAs; and blue, rRNAs. Inner and outer circles represent genes oriented on major and minor strands, respectively.
The M. vitrata (New World) mitogenome showed a high A+T content 80.70% (A = 40.30%, T = 40.4%, G = 7.99%, and C = 11.31%) (Table 2), and this is consistent with the high A+T content of insect (Clary and Wolstenholme, Reference Clary and Wolstenholme1985; Crozier and Crozier, Reference Crozier and Crozier1993) and lepidopteran mitogenomes (Liu et al., Reference Liu, Li, Pan, Dai, Zhu, Lu and Xiang2008). The nucleotide composition of the PCGs and the AT- and GC-skew calculated for all available crambid mtDNA genomes are presented in Table 2. The average AT-skew of the crambid mitogenomes was 0.006, ranging from −0.024 to 0.03, whereas the average GC-skew was −0.195, ranging from −0.26 to −0.17. The AT-skew of M. vitrata (New World) was −0.002. A similar negative AT-skew was also previously reported for C. medinalis (Chai et al., Reference Chai, Du and Zhai2012) and Spoladea recurvalis (He et al., Reference He, Zou, Zhang, Ma, Zhang and Yue2015) (Table 2). This indicates a bias for T over A nucleotides. Consistent with other lepidopteran mitogenomes that exhibit negative GC skewness, the GC-skew of M. vitrata (New World) was −0.17, indicating a bias for C over G nucleotides.
Table 2. Nucleotide composition in the whole mitogenomes of Maruca vitrata (New World) and other available mitogenomes from six additional species from the lepidopteran Family Crambidae
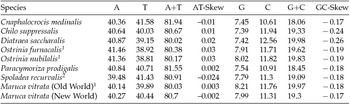
1 Partial mitogenome sequences that lacked portions of the AT-rich control region sequence.
2 Partial mitogenome sequences for which the cox1 gene sequence is publicly unavailable.
Protein coding genes
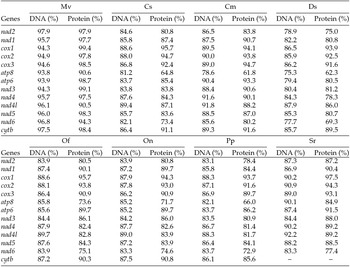
The putative start codons of PCGs used by M. vitrata (New World) (ATN; Table 1) were identical to those previously known in animal mitogenomes (Wolstenholme, Reference Wolstenholme1992). CGA was used as the start codon for cox1 as previously reported for lepidopterans such as Bombyx mori (Yukuhiro et al., Reference Yukuhiro, Sezutsu, Itoh, Shimizu and Banno2002), O. nubilalis and O. furnacalis (Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005), S. recurvalis (He et al., Reference He, Zou, Zhang, Ma, Zhang and Yue2015), Adoxophyes honmai (Lee et al., Reference Lee, Shin, Kim, Park, Cho and Kim2006), C. raphaelis (Kim et al., Reference Kim, Lee, Seol, Yun, Lee, Hwang and Jin2006), Antheraea pernyi (Liu et al., Reference Liu, Li, Pan, Dai, Zhu, Lu and Xiang2008), B. mandarina (Pan et al., Reference Pan, Yu, Xia, Dai, Liu, Lu, Zhang and Xiang2008), Ochrogaster lunifer (Salvato et al., Reference Salvato, Simonato, Battisti and Negrisolo2008), Artogeia melete (Hong et al., Reference Hong, Jiang, Yu, Yang, Li, Xue and Wei2009), Eriogyna pyretorum (Jiang et al., Reference Jiang, Hong, Yu, Li, Yang, Liu and Wei2009), Hyphantria cunea (Liao et al., Reference Liao, Wang, Wu, Li, Zhao, Huang, Niu, Liu and Li2010) and M. vitrata (Old World) (Margam et al., Reference Margam, Coates, Hellmich, Agunbiade, Seufferheld, Sun, Ba, Sanon, Binso-Dabire, Baoua, Ishiyaku, Covas, Srinivasan, Armstrong, Murdock and Pittendrigh2011b ). The termination codon TAA was used in all PCGs with the exception of nad3, which had TAG. Also, consistent with some other insect mitogenomes, cox1 and cox2 both had incomplete (T–) stop codon (Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005; Salvato et al., Reference Salvato, Simonato, Battisti and Negrisolo2008) (Table 1). The presence of incomplete stop codons is a molecular feature shared with all lepidopteran mitogenomes sequenced to date (Brower, Reference Brower1994; Yukuhiro et al., Reference Yukuhiro, Sezutsu, Itoh, Shimizu and Banno2002; Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005; Kim et al., Reference Kim, Lee, Seol, Yun, Lee, Hwang and Jin2006; Lee et al., Reference Lee, Shin, Kim, Park, Cho and Kim2006; Cameron and Whiting, Reference Cameron and Whiting2008; Hong et al., Reference Hong, Lee, Jo, Park, Kim, Hwang, Jin, Kang, Kim, Han and Kim2008) and with many arthropod mitogenomes (Boore, Reference Boore1999). The motif TTAG has been observed immediately upstream of the putative cox1 CGA start codon in some lepidopteran mitogenomes and has been proposed to serve in non-standard initiation (Yukuhiro et al., Reference Yukuhiro, Sezutsu, Itoh, Shimizu and Banno2002). We observed the motif ATAG immediately upstream of the CGA start codon of the M. vitrata (New World) cox1 gene. This motif is conserved in some lepidopteran mitogenomes and lacking in others (Margam et al., Reference Margam, Coates, Hellmich, Agunbiade, Seufferheld, Sun, Ba, Sanon, Binso-Dabire, Baoua, Ishiyaku, Covas, Srinivasan, Armstrong, Murdock and Pittendrigh2011b ). Among the available crambid mitogenomes, O. furnacalis and O. nubilalis both have the motif TTAG upstream of the start codon of the cox1 gene, while M. vitrata (Old World) has ATAG. Reduction in mitogenome size has led to minimal intergenic space as well as overlap between adjacent genes in M. vitrata (New World). Atp8 and atp6 PCGs had a seven-nucleotide overlap (Table 1). This feature is common to all lepidopteran mitogenomes known (Brower, Reference Brower1994; Yukuhiro et al., Reference Yukuhiro, Sezutsu, Itoh, Shimizu and Banno2002; Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005; Kim et al., Reference Kim, Lee, Seol, Yun, Lee, Hwang and Jin2006; Lee et al., Reference Lee, Shin, Kim, Park, Cho and Kim2006; Cameron and Whiting, Reference Cameron and Whiting2008; Hong et al., Reference Hong, Lee, Jo, Park, Kim, Hwang, Jin, Kang, Kim, Han and Kim2008) and is found in many animal mitogenomes (Boore, Reference Boore1999). The A+T content of the PCGs was 79.4%, which is lower than the A+T content of the mitogenome as a whole (80.7%). The total number of codons on the majority strand, excluding stop codons, was 3,777 and showed a strong bias toward AT-rich codons, with the four most prevalent codons being Leu, UAA (481); Ile, AUU (443); Phe, UUU (344); and Met, AUA (267) (Table 3). The codon usage prevalence was consistent with that found in M. vitrata (Old World). Comparison of the 13 PCGs across the eight available crambid mitogenomes with those of M. vitrata (New World) resulted in 80.4% and 99.1% identity at the nucleotide level, and 61.8% and 99.4% identity at the amino acid level (Table 4). At both nucleotide and amino acid levels, all the 13 PCGs of M. vitrata (New World) showed the highest identity with those of M. vitrata (Old World).
Table 3. Codon usage and relative synonymous codon usage (RSCU) in Maruca vitrata (New World) mitogenome
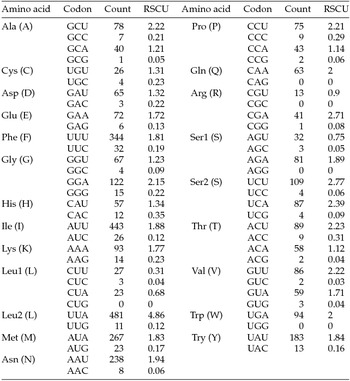
Table 4. Comparison of the percent nucleotide and amino acid identity in the 13 protein coding gene sequences of Maruca vitrata (New World) with orthologs in 6 other crambid lepidopteran mitogenomes [Mv – M. vitrata (Old World), Cs (C. suppressalis), Cm (C. medinalis), Ds (D. saccharalis), Of (O. furnacalis), On (O. nubilalis), Pp (Paracymoriza prodigalis), Sr (Spoladea recurvalis)]; coxI gene sequence not available)
Transfer and ribosomal RNA genes
The published mitogenome of M. vitrata (Old World) is incomplete and thereby has 19 tRNAs and a part of the rRNA reported. However, the M. vitrata (New World) mitogenome encoded the 22 tRNAs typically found in animal mitogenomes (Boore, Reference Boore1999) and all formed the canonical cloverleaf-like tRNA secondary structure, with the exception of trnS1, in which its dihydrouridine (DHU) arm formed a simple loop. The lack of DHU arm in trnS1 is a common condition in metazoan mitogenomes (Taanman, Reference Taanman1999) but is not found in all lepidopterans. For example, A. honmai has all the tRNAs with a complete clover leaf structure (Lee et al., Reference Lee, Shin, Kim, Park, Cho and Kim2006). Of the 22 tRNA genes, 14 genes were encoded on the majority strand, while eight were encoded on the minority strand. The length of tRNAs ranged from 63 bp to 71 bp. Consistent with what is observed in all other insect mitogenomes, the mitogenome of M. vitrata (New World) contained two rRNAs with a total length of 2,069 bp. The large rRNA (rrnL) had a length of 1,304 bp, while the small rRNA (rrnS) had a length of 765 bp (Table 1).
Non-coding regions
The M. vitrata (New World) mitogenome contained characteristics typical of other lepidopteran mitogenomes. The 7 bp intergenic spacer located between trnS2 and nad1 contained the ATACTAA motif, which is conserved across the order Lepidoptera (Cameron and Whiting, Reference Cameron and Whiting2008), including M. vitrata (Old World). This motif is possibly fundamental to site recognition by the transcription termination peptide (mtTERM protein) (Taanman, Reference Taanman1999) and is present in most insect mitogenomes (Cameron and Whiting, Reference Cameron and Whiting2008). Insect mitogenomes often contain a non-coding A+T-rich region that varies considerably in length among insect species, or even within the same species (Zhang and Hewitt, Reference Zhang and Hewitt1997). Consistent with findings from other insects, the largest non-coding region was the A+T-rich control region, although it varies in size in crambids (Hua et al., Reference Hua, Li, Dong, Cui, Xie and Bu2008; Oliveira et al., Reference Oliveira, Raychoudhury, Lavrov and Werren2008; Ma et al., Reference Ma, Liu, Yang and Kang2009). Tandem repeat sequences are common in the control region of insects and can vary in length and copy number (Dotson and Beard, Reference Dotson and Beard2001). Repeats in Lepidoptera share greater degrees of conservation than in other insect groups (Cameron and Whiting, Reference Cameron and Whiting2008). The A+T rich control region in M. vitrata (New World) was estimated at 341 bp and contained (AT) tandem repeats. The A+T-rich region contained a conserved ATAG motif followed by an 18 bp poly-T stretch. This feature has been found in C. medinalis, C. suppressalis and D. saccharalis, but the length of the poly-T stretch varies between species. This structure is suggested to function as a signal for mitochondrial DNA replication initiation (Hu et al., Reference Hu, Zhang, Hao, Huang, Cameron and Zhu2010; Yin et al., Reference Yin, Hong, Wang, Cao and Wei2010; Kim et al., Reference Kim, Wan, Kim, Hwang and Kim2010). A poly-A stretch commonly observed in other lepidopteran mitogenomes was also found immediately upstream of trnM in M. vitrata (New World). The (AT)4, (AATT)4 and (AT)9 repeats in the M. vitrata control region were predicted (Supplementary Table 2) of which (AT)9 was similar to an (AT)8 in Hyphantria cunea (Liao et al., Reference Liao, Wang, Wu, Li, Zhao, Huang, Niu, Liu and Li2010) and (AT)7 in Ochrogaster lunifer (Salvato et al., Reference Salvato, Simonato, Battisti and Negrisolo2008).
Comparative genome analysis and estimation of divergence times
Protein coding sequence was shown to be highly similar between M. vitrata (New World) and M. vitrata (Old World) mitogenomes (see above; Table 4). Comparison between M. vitrata (New World) and M. vitrata (Old World) coding sequences found 476 nt substitutions in the 11,250 nt consensus alignment (4.23% divergence), of which 74 (15.5%) were non-synonymous mutations. This suggests that purifying selection may be curbing the rate of amino acid change in Maruca. Novel non-synonymous mutations were directly shown to be under negative selection in haplotypes derived from mutator strains of mice (Stewart et al., Reference Stewart, Freyer, Elson, Wredenberg, Cansu, Trifunovic and Larsson2008) such that the accumulation of mildly deleterious mutations might lead to reduced fitness and result in removal from the population. The alignment of entire mitogenome sequences from M. vitrata (Old World) and M. vitrata (New World) resulted in a 14,039 consensus (gaps excluded) and showed a total of 526 nucleotide differences (3.75%) (Supplementary Table 3). Making a simple assumption of a clock-like rate of 2% change per million years estimated from the Drosophila species group (Powell et al., Reference Powell, Caccone, Amato and Yoon1986) and with an effective rate of change of 1.15% per million years for Lepidoptera (Brower, Reference Brower1994) and acknowledging the potential that variance in this rate may have affected the degree of haplotype divergence among Maruca mitochondrial genomes, the two strains may have diverged ~1.83 to 2.12 mya. These estimates likely suffer from uneven rates across taxonomic groups and time (Ho and Lo, Reference Ho and Lo2013). Regardless, evolutionary rates have been estimated from calibrated molecular clocks (Papadopoulou et al., Reference Papadopoulou, Anastasiou and Vogler2010) and have resulted in estimates similar to the standard rate (Brower, Reference Brower1994). With lack of fossil evidence to accurately calibrate the accumulation of substitutions across the lepidopteran mitochondrial genome lineages, the derivation of an accurate divergence time estimate between M. vitrata strains remains elusive and beyond the scope of this study.
Conclusion
The 15,385 bp mitogenome of M. vitrata (New World) has a size and gene order typical of mitogenomes from the insect order Lepidoptera. The aim of this study was achieved by defining the previously undetermined levels of mitochondrial genome variation between strains of M. vitrata. In doing so, a substitution bias at non-synonymous sites was defined, which is typical for highly conserved mitochondrial PCG, including the previously published mitogenome coxI by Margam et al. (Reference Margam, Coates, Hellmich, Agunbiade, Seufferheld, Sun, Ba, Sanon, Binso-Dabire, Baoua, Ishiyaku, Covas, Srinivasan, Armstrong, Murdock and Pittendrigh2011b ). This short-term change identified here provides valuable information regarding variation between geographically distinct strains of a species that is analogous to that shown by comparisons between other closely related insect species (Coates et al., Reference Coates, Sumerford, Hellmich and Lewis2005; Coates, Reference Coates2014). Specifically, these whole mitochondrial genome analyses provide estimates of divergence time and short-term patterns of sequence evolution that are more accurate compared to the use of single mitochondrial gene sequence data. Although not explored in the current study, the rapid de novo assembly of mitogenomes from NGS sequence data presents future opportunities for phylogenomic studies within and among species of Lepidoptera.
Acknowledgements
This project has been made possible through support provided by the Legumes Innovation Lab, formerly known as the Dry Grains Pulses Collaborative Research Support Program (CRSP), by the Bureau for Economic Growth, Agriculture, and Trade, US Agency for International Development, under the terms of Grant No. EDH-A-00-07-00005. The United States Department of Agriculture, Agricultural Research Service (USDA-ARS: CRIS Project 3625-22000-017-00) and the Iowa Agriculture 567 and Home Economics Experiment Station, Ames, IA (Project 3543) supported a portion of research data collection and analysis. TAA was supported by International Student Research Fellowship from the Howard Hughes Medical Institute, Maryland, USA.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S1742758417000157.







 Open access
Open access