



Introduction
Multiple clinical trials have failed to identify the master key to improve the outcome of patients suffering from traumatic brain injury (TBI). One of the challenges is that not all TBI are created equal and not all secondary neuronal injury have the same underlying pathophysiology.
The increasing use of invasive intracranial monitoring, including electrocorticography (ECoG), has enabled the discovery of cortical spreading depolarization (CSD) in a significant proportion of TBI. Reference Soldozy, Sharifi and Desai1 In fact, a recent study showed that over 50% of TBI requiring surgical intervention had evidence of CSD. Reference Soldozy, Sharifi and Desai1 The biomechanical force associated with TBI leads to cortical shear stress that results in microvascular injury. If the shear stress surpasses, a certain threshold depolarization ensues within the injured area. A significant amount of depolarization may trigger CSD, potentially contributing to the symptoms associated with TBI and influencing prognostic and outcome.
CSD is a pathologically triggered wave that propagates slowly through the cerebral gray matter, leading to an excess of intracellular calcium, cytotoxic edema, and release of excitatory neurotransmitters implicating, among others, the N-methyl-d-aspartate receptor signaling (NMDA). Reference Hartings, Shuttleworth and Kirov2–Reference Marrannes, Willems, De Prins and Wauquier4 The implication of CSD in the secondary neuronal injury has been well documented in recent literature. CSD seems to be the underlying process explaining the rare occurrence of “migraine strokes” in relation to prolonged aura. The presence of lesional CSD is well documented by the Co-Operative Studies on Brain Injury Depolarizations (COSBID) group in various neurological insults, including subarachnoid hemorrhage, stroke, and hemiplegic migraines. Recent literature suggests the presence of CSD in a various array of CNS insults including the recent hypothesis that it may be the underlying driving factor in transient global amnesia Reference Spiegel, Smith and Wade5 and in certain cases of delirium. Reference Nielsen, Urdanibia-Centelles and Vedel-Larsen6
CSD, originally described and recognized in migraine aura, has since been studied in a vast array of neurological entities, and research on its pathophysiological role and prognostic impact has gained interest in recent years. However, the literature regarding its role in chronic subdural hematoma (cSDH) remains scarce. In addition, most research have focused on the pathophysiological process of CSD, a normal first step in describing an entity, but very little literature exists linking the electrochemical process of CSD to its clinical picture for everyday clinicians.
In practice, clinicians are frequently confronted to transient focal neurological symptoms (TNS). The differential diagnosis evoked in such patients includes transient ischemic accidents, seizures, fluctuating mass effect, and CSD. Although CSD is a recognized cause of TNS, it remains a neglected and underdiagnosed etiology despite its significant prevalence and importance.
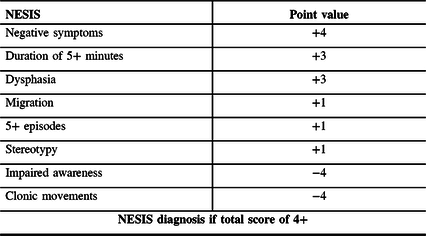
Until recently, there was no direct evidence of the existence of CSD in cSDH. Recently, the term Non-Epileptical Stereotypical Intermittent Symptoms (NESIS) was proposed to describe a subgroup of patients presenting with TNS, unlikely to be of convulsive or ischemic nature, in the context of cSDH. An experimental clinical bedside weighted scoring system was proposed to screen for NESIS patients with TNS unlikely to be of convulsive etiology (Table 1). Reference Levesque, Iorio-Morin, Bocti, Vézina and Deacon7
Table 1: Proposed scoring system for the diagnosis of NESIS
Chronic Subdural Hematoma
cSDH are frequently encountered in clinical practice with an estimated incidence approaching 20/100,000 patient-year. Reference Iorio-Morin, Touchette, Lévesque, Effendi, Fortin and Mathieu8 Patients presenting with cSDH may show a myriad of symptoms that may guide therapeutic decisions.
A significant proportion of patients presenting with cSDH may show a fluctuating course of TNS. Such TNS may be encountered upon initial presentation, during the course of the hospitalization or upon subsequent follow-up. Most of them will receive a presumptive diagnosis of acute symptomatic seizures or epilepsy and will be treated as such. Epilepsy has been reported to occur in as many as 25% of patients post-SDH evacuation. Reference Rabinstein, Chung, Rudzinski and Lanzino9 However, a significant subgroup of patients with cSDH will present with a distinct clinical course that may include prolonged negative symptomatology, unfavorable response to antiepileptic drugs (AED), and a negative workup including electroencephalography (EEG), all atypical for seizure activity. This raises a diagnostic dilemma and suggests the potential presence of an alternative causal etiology for their TNS. A small recent retrospective study performed in a cohort of 84 patients with cSDH revealed an incidence of TNS of 26%, of which 42% remained with an inconclusive diagnosis. Reference Douri, Levesque, Mathieu and Iorio-Morin10
TBI results in a neuroinflammatory response leading to the release of immune mediators. Injured cells secrete damage-associated molecular particles which interact with receptors on immune cells contributing to an influx of inflammatory cytokines which activate microglia. The microglia are then implicated in both tissue repair and neurodegeneration. This inflammatory environment may contribute to promoting CSD. Reference Soldozy, Sharifi and Desai1
The increased intracranial pressure caused by SDH may decrease the cerebral perfusion pressure (CPP) and increase the microvasculature resistance, thereby lowering the brain blood volume, in accordance to Poiseuille’s law. This process creates a supply–demand energy mismatch potentially contributing to secondary neuronal injury. A vicious circle may ensue leading to increased energy demands and decrease in supply due to inappropriate compensatory mechanism in the injured brain parenchyma. This whole paradigm underlies many essential pathophysiological processes known to enable and sustain CSD.
Cortical Spreading Depolarization
CSD, as originally defined in 1944, represents the process of imbalance in transmembrane ionic gradient in neuronal cells leading to an autopropagating wave spreading contiguously in the gray matter, irrespective of vascular territories, at a speed of 1–9 mm/min. The majority of migraine aura are visual in nature due to the lower ratio of astrocytes in Brodman area 17, which are known to be protective and inhibitory against CSD. Reference Torrente, Cabezas, Avila, García-Segura, Barreto and Guedes11,Reference Fabricius, Akgoren and Lauritzen12 Astroglial cells have the capacity to buffer potassium and glutamate, thus preventing CSD generation and propagation. An increased sensitivity to CSD has been shown with astroglial pathology. Reference Largo, Herreras and Ibarz13
Since its original description by Laeo and Lashley in 1944, the understanding of the underlying pathophysiology of CSD and its existence in various neurological entities has been growing considerably. Significant advances in the field were rendered possible by the works of Strong, Sackowitz, Hartings, and COSBID group, among others, that have paved the way to a better understanding of CSD. Reference Dreier, Fabricius and Ayata14
The energy demand of CSD is estimated to exceed by a factor of 4–8 that of a focal seizure. This high energy demand must be matched by the energy supply in order to allow the phase of repolarization to occur. Transient electrocerebral inactivity, during which neurons can no longer generate action potentials, classically follows CSD until repolarization of the cellular membrane is complete. In the healthy brain, this energy demand is quickly compensated by healthy compensatory energy mechanism leading to short lasting symptomatology. Electrochemically, CSD is characterized by an influx of calcium and sodium and an efflux of excitatory neurotransmitters which are buffered by the astrocytes through their role in glutaminergic recapture. This exchange leads to an elevated lactate-to-pyruvate ratio, decreasing glucose, pH, and activating the anaerobic pathway, supporting the implicated ischemic pathophysiology. Reference Parkin, Hopwood and Jones15 The electrochemical imbalance that results alters the cerebral vasoreactivity. On a neurovascular level, an initial phase of hyperemia, corresponding to the depolarization phase, is followed by transient oligemia until repolarization is complete. In the healthy brain, this oligemia leads to subsequent compensatory vasodilatation (induced by the release of nitric oxide and arachidonic acid) and is followed by compensatory hyperemia. Reference Feuerstein, Backes and Gramer16–Reference Piilgaard and Lauritzen18
However, these compensatory mechanisms may be deficient in the injured brain and isoelectric (or perilesional) cortical depolarization may ensue if the lesional tissue cannot match the supply–demand mismatch created by the CSD. In addition, the injured brain may react pathologically to the energy supply–demand mismatch with paradoxal vasoconstriction further increasing the mismatch, reducing the oxygen supply, and potentially contributing to propagating neuronal damage. This paradoxal cascade of events limits the ability of the transcellular membranes to repolarize which extends the period of electrocerebral silence and symptomatology. Reference Lauritzen, Dreier, Fabricius, Hartings, Graf and Strong19,Reference Lauritzen and Strong20 It has been shown that CSD duration is inversely proportional to systemic arterial pressure supporting the importance of cerebral perfusion pressure. Reference Sukhotinsky, Yaseen and Sakadžić21 The existence of CSD supports the presence of underlying residual viable and potentially salvageable neuronal tissue, suggesting the presence of a potential treatment target which could potentially improve patients’ outcomes.
This pathological response to CSD has been shown to have significant prognostic impact in various studies. A recent study using electrocorticography in patients with TBI has revealed an incidence of CSD of 60.1%. CSD was associated with lower prehospital systolic blood pressure and a poor neurologic recovery (odds ratio [OR] 2.29). Reference Hartings, Bullock and Okonkwo22,Reference Pacheco, Hines-Lanham and Stratton23 This electrochemical and neurovascular imbalance explains the frequently observed differences in clinical presentation of lesional CSD symptomatology versus the more benign nonlesional CSD, such as that observed in migraine aura. Reference Hartings, Andaluz and Bullock24 The incidence of CSD has been reported to be as high as 60% in TBI, 80% in SAH, and over 90% in aggressive strokes. Reference Dreier, Fabricius and Ayata14 A recent animal study revealed that the presence of CSD correlated with concussive symptomatology post-mild TBI and a higher number of apoptic cells. Reference Taş, Solaroğlu and Gürsoy-Özdemir25 Human studies have shown a correlation between the presence of CSD and post-injury behavior. Reference Chung, Oka and Ayata26 Delayed cerebral ischemia after SAH is associated with upward of 50% mortality and is the most feared secondary complications in initial survivors of SAH. Initially attributed to vasospasm, recent data suggest that it is rather caused by a complex synergic mix of mechanisms of which CSD seems to be the driving factor. Nimodipine, a calcium channel blocker, has been shown to reduce the incidence of delayed cerebral ischemia despite having no impact on the vasospasm itself. Animal literature and human evidence on migraineurs have shown calcium channel blockers to reduce the incidence of CSD by its stabilizing effect on the neuronal membrane potential, thus suggesting its effect on delayed cerebral ischemia may be related to CSD inhibition rather than on vasospasm. Reference Bouley, Chung, Ayata, Brown and Henninger27–Reference Zheng, Schoell, Sanchez-Porras, Diehl, Unterberg and Sakowitz30 Cortical SAH is characteristic of amyloid angiopathy and associated with “amyloid spells” which represent the TNS often associated with the cortical SAH or superficial siderosis. These areas of superficial siderosis are particularly characteristic of amyloid angiopathy.
Experts agree that not all CSD are harmful. The distinction between benign CSD, such as that observed in most migraine aura, and harmful CSD, such as that generally described in lesional CSD, must be made. Further blurring the margins between benign and damaging CSD is the belief that some CSD may potentially be beneficial by contributing to synaptic strengthening, neurotrophic factor stimulation, neuroprotection preconditioning, neurogenesis stimulation, and hyperemic response. Reference Shuttleworth, Andrew and Akbari31
Multiple animal studies have shown promising therapeutic options to target the pathophysiology of CSD, including arginine, lamotrigine (LTG), verapamil, flunarizine, ketamine (NMDA antagonist), magnesium, and topiramate (TPM), all likely due to their stabilizing effect on the neuronal cellular membrane and on specific receptors. Reference Klass, Sánchez-Porras and Santos32 All but arginine have an effect as calcium channels blockers. Interestingly, calcium channel blockers are among the medication approved for hemiplegic migraines where in the motor manifestations seem to be a reflection of prolonged CSD. Genetic forms of migraine are associated with mutations in sodium, potassium, and calcium channels which are associated with an increased sensitivity and decreased threshold to CSD triggering and a tendency to repeated bouts of CSD after a single stimulus. Reference van den Maagdenberg, Pizzorusso and Kaja3 Those specific medications appear to inhibit the spread of the depolarization wave by stabilizing the electrochemical membrane, thereby helping restore the physiological gradient through mechanisms which include sodium channel, calcium channel, and glutaminergic interactions. Among the studied medications, TPM and ketamine appear to show the most consistent and reproductive effects on CSD ablation. Reference Lampl, Katsarava, Diener and Limmroth33–Reference Sakowitz, Kiening and Krajewski37 In addition, the literature has demonstrated the important impact of hypotension, hypoglycemia, and hyperthermia in the pathophysiology triggering and sustaining CSD, and it is therefore advised to attempt aggressive normalization of those hemodynamic parameters before initiating any subsequent treatment.
The question of what to do when facing CSD remains elusive. The literature concerning CSD has been growing in recent years and yet no study has investigated whether interventions targeting the suppression of CSD truly have clinical impact on patient outcomes. A group of experts from the international conference on spreading depolarizations (iCSD) have recently published a statement regarding the optimal approach to preconize regarding CSD. Ketamine, an NMDA receptor antagonist, is the suggested leading treatment option to suppress CSD based on published results from multiple centers and expert opinion. Dilemma remains regarding which CSD should be treated and if treatment impacts prognostic outcome. It appears that ketamine could suppress CSD in sites remote from the vulnerable area, wherein CSD could potentially have a beneficial effect, more so than in the ischemic area, which further complexifies treatment decision. Currently, there is no existing consensus on treatment approach for such patients. Reference Helbok, Hartings and Schiefecker38
Cortical Depolarization in Subdural Hemorrhage: Connecting the Dots
Although CSD’s presence and pathophysiological role have been recognized in a growing array of CNS insults, its presence was, until recently, not confirmed in the SDH population.
A single-center retrospective study recently investigated the semiology of patients presenting with transient neurological symptoms post-SDH evacuation. Reference Levesque, Iorio-Morin, Bocti, Vézina and Deacon7 Significant discrepancies were observed between those likely to suffer from a convulsive etiology and a distinct subgroup of patients, defined by the term NESIS. Based on the observed differences, a provisional clinical score (Table 1) was proposed to screen for the patients whose TNS were unlikely to be attributable to seizures. The score attributes points weighted based on a review of the evidence in the literature and the results of the retrospective study. In order of decreasing point value, the score adds points for the presence of negative symptomatology, prolonged duration of symptoms, dysphasia, migrational characteristics to the symptoms, repeated and stereotyped symptoms, and subtracts points for the presence of impaired awareness and clonic movements. Of particular interest, the NESIS group showed significantly better outcomes, despite having more prolonged and frequent TNS and being significantly more refractory to standard antiepileptic treatment. This paradox in semiology, therapeutic, and prognostic response supports the existence of a distinct pathophysiology than epilepsy. Despite being limited by its retrospective design and the intrinsic limitations of scalp electroencephalography regarding its sensitivity in detecting epilepsy, this study describes a new clinical syndrome with strong clinical implications and proposed an easy-to-use bedside clinical score to help detect it. They have hypothesized that NESIS could be the clinical representation of underlying CSD.
A subsequent retrospective analysis of consecutive patients hospitalized for cSDH from the same center was performed. In the cohort of 84 patients, 26% experienced TNS of whom 59% scored positive for NESIS based on the score proposed in the original study. Reference Douri, Levesque, Mathieu and Iorio-Morin10 Mortality rate was of 0% in the NESIS group compared to 33% in the non-NESIS group, once again supporting a more favorable prognostic in the NESIS than non-NESIS group of patients with cSDH and TNS. Although limited by the small population and the retrospective design, those results strengthen the separation of two distinctive groups of patients which can be separated by the proposed NESIS score. The distinction between NESIS-positive and NESIS-negative patients among the population of patients with TNS in the context of cSDH seems to have significant prognostic and therapeutic implications. Interestingly, only 14% of NESIS patients responded to phenytoin, levetiracetam (LEV), or lacosamide compared to 100% of non-NESIS TNS. In addition, 100% of the NESIS group responded to a switch from their original AED toward TPM or LTG, both medications with proven efficacy to suppress CSD. Those results are of particular interest because they offer strong arguments supporting the existence of NESIS, its significant prevalence in cSDH, and the potential value of the proposed clinical score in screening for it. In addition, the response to AED known to have therapeutic effect on CSD may provide indirect arguments supporting that CSD may be the underlying pathophysiology of TNS in NESIS patients.
A recent study using ECoG in 40 patients with chronic SDH post-surgical evacuation revealed the presence of definite CSD in 15% of patients post-surgical evacuation (independent of the presence or absence of TNS). Reference Mohammad, Abbas and Shuttleworth39 One of the cases demonstrated a time-lock focal neurological deterioration at the onset of CSD clusters with no alternative causal explanation on MRI or EEG supporting that his TNS may have been directly caused by the CSD. The presence of CSD proved to be of prognostic significance and was associated with postoperative neurological deterioration in 50% compared to 8.8% in the subgroup without evidence of CSD. This study is pivotal in confirming the presence of CSD in patients with chronic SDH and in demonstrating their causative role in a subset of TNS in that population. Those results, combined with the NESIS study, support that CSD may be the pathophysiology driving otherwise unexplained fluctuating neurological deterioration in a significant subgroup of patients with cSDH.
Discussion
CSD can be triggered by any neurological insult irritating cortical neurons and starting the self-propagating wave of depolarization. Injured tissue has a limited capacity to respond and compensate to the energy demand associated with CSD and necessary to stop it and enable repolarization contributing to prolonged symptomatology, lesional extension, and prognostic implications.
Despite the recognition of CSD in a growing array of neurological entities, its presence in SDH clinical literature remains limited. The absence of a clinical tool to help recognize it likely contributes to an underrecognition of the syndrome in everyday clinical practice. Most current research has revolved around the pathophysiological basis of CSD which, although a necessary initial step, may appear as a more abstract process to clinicians in everyday practice. Recent research attempts to answer some of these gaps between the literature and clinical practice. A series of 15 cases published in 1992 described patients with a semiology reminiscent of NESIS, but did not propose diagnosis hypothesis at the time. All had negative EEG and only three had significant atherosclerosis in the symptomatic territory. Upon analysis of this series of case, a significant proportion of the patients described would have been characterized as NESIS according to the proposed clinical score. Reference Kaminski, Hlavin, Likavec and Schmidley40 Another limitation is the fact that some TNS might also have been caused by epileptic seizures that were missed on scalp-EEG or missed because of the lack of prolonged EEG monitoring. Despite this limitation, the proportion of negative EEG, despite most being completed near the seizure event and many patients having multiple EEG, far surpasses what would be expected if all of the events were related to seizures. In addition, the distinct semiology, therapeutic response, and prognosis between both groups support the existence of a distinct underlying pathophysiology in the NESIS group.
The presence of CSD in various neurological entities may often be missed and seems to be underestimated. Reference Bapteste, Marinesco and Lieutaud41 CSD should be an integral part of the differential diagnosis to evoke when facing TNS in any neurological population. Growing literature on CSD will likely lead to further development in the recognition of CSD by clinicians and clearer distinctions between which CSD merits aggressive treatment and which do not. CSD represents a new therapeutic target in a subset of patients with fluctuating neurological deficits, and optimal targeted therapeutic approach remains unclear until further prospective trials on the matter are conducted.
A significant challenge in the near future remains in that CSD cannot be reliably diagnosed noninvasively. Reference Drenckhahn, Winkler and Major42 While ECoG studies are desirable to further our understanding of pathophysiology, routine invasive monitoring would add unnecessary complexity if systematically performed in all cSDH cases. Given that the preliminary data on CSD in cSDH seem to suggest it may be a benign marker compared to epilepsy and that both can be treated by oral AEDs, empirical treatment could be reasonable. To that end, the NESIS score could be sufficient to clinically identify cSDH patients experiencing CSD and treat them without prior invasive monitoring studies. Assuming the validity of that score can be confirmed and replicated in a distinct population, it could also prove useful in future prospective trials. Reference Iorio-Morin and Levesque43 The proposal of NESIS and the value of its clinical score should be validated and replicated. At the moment, the score has only been studied in patients suffering from TNS in relation to SDH and, until studied in distinct populations, it should not be directly extrapolated to other cases of TNS or suspected lesional CSD. Reference Levesque, Iorio-Morin, Bocti, Vézina and Deacon7 Although the fact that NESIS represents the manifestation of CSD remains hypothetical, there seems to be a growing array of evidence in the literature to support that statement. It is also important to note that CSD and seizures are not always mutually exclusive and both phenomenologies could happen in the same patient. Interestingly, CSD seems to present differently in irritative etiology versus intrinsic neuronal injuries. As such, a majority of CSD in migraine aura or in cortical SAH or superficial siderosis present with positive migrational symptomatology, whereas CSD in cSDH, stroke, and aneurismal SAH seems to present with negative symptoms in a majority of cases.
Interestingly, the presence of CSD in patients with SDH proved to be a marker for worse prognostic outcome, Reference Mohammad, Abbas and Shuttleworth39 while the presence of NESIS, believed to be a marker of CSD, was associated with favorable outcome in two distinct studies. Reference Levesque, Iorio-Morin, Bocti, Vézina and Deacon7,Reference Douri, Levesque, Mathieu and Iorio-Morin10 Although the different study design limits the possibility for direct comparisons, such discrepancies could be related to the different population studied. The NESIS research studied only patients with TNS, hence comparing the prognosis of NESIS versus seizures, whereas the prospective trial studied all patients with SDH, independent of the presence or absence of TNS. With a lot of limitations, this could indirectly suggest that the presence of CSD could be intermediate between the absence of any abnormal electrophysiological phenomena (including CSD) and the presence of seizures as a prognostic marker. As such, future studies in the field could reveal the presence of three distinct subgroup of patients with different prognostic values.
Indirect evidence strongly suggests NESIS to be a manifestation of CSD and, if validated, the replication of the proposed NESIS score to screen for the presence of CSD could prove useful in both research setting and clinical practice. Future investigations are needed to validate the score and see if it could be extrapolated and applied to lesional CSD from other etiologies than SDH. Reference Adam, Levesque, Deacon and Iorio-Morin44
Generating evidence for NESIS (GENESIS) is a planned, prospective trial that will study the population of patients presenting with TNS in the context of cSDH with a positive NESIS score by randomizing them into an LEV and a TPM group. Both medications have similar efficiency in treating seizures, although LEV has gained popularity compared to TPM due to its favorable side effect profile and tolerability. The effect of LEV on CSD seems to be negligible in animal studies, whereas TPM has been shown to have an incisive effect on aborting CSD. Reference Lin, Hsu and Cheng45 The study aims to show discrepancy in the evolution of patients in both groups, which would support the presence of a distinct etiology to epilepsy and the value of the NESIS score in detecting it while providing strengthening arguments supporting the statement that NESIS is the manifestation of CSD and providing evidence to potential therapeutic avenues and their potential prognostic impacts on patient outcomes.
Conclusion
CSD is well recognized in various neurological insults and evidence is cumulating on its prognostic impact. Questions remain regarding if treating CSD can impact outcome in patients and if so which patients would benefit most from an aggressive treatment approach. Future studies are required in order to provide evidence on how to recognize and optimally treat CSD in patients suffering from it. Among patients presenting with cSDH associated with TNS, a subpopulation exists in which the symptoms are unlikely to be epileptic in nature. The term NESIS has been proposed, and growing evidence suggest it could be attributable to underlying CSD. The definition and recognition of NESIS could have significant diagnostic, therapeutic, and prognostic implications.
Many questions remained unanswered, and the optimal approach to diagnosing and treating NESIS remains speculative opening doors to this interesting field of research in hopes of better tailoring treatment approach and improving patients’ outcomes.
Disclosures
None.
Statement of Authorship
ML: Review of literature and writing of the manuscript; Paper concept, acquisition of data and paper submission. CI-M, SA, CD: Revision of the manuscript.

