


Drug use disorders (DUDs) are complex traits influenced by a variety of environmental factors and genetic risks. Depending on the substance, genetic risks account for 40–70% of the liability to drug initiation (DI) and DUDs (Kendler & Prescott, Reference Kendler and Prescott2006) with increasing heritability as individuals progress to DUDs (Kendler et al., Reference Kendler, Chen, Dick, Maes, Gillespie, Neale and Riley2012). The remaining variance can be predicted by a variety of social-environmental risks often identifiable in early to mid-childhood (Caspi et al., Reference Caspi, Moffitt, Newman and Silva1996; Casswell et al., Reference Casswell, Pledger and Pratap2002; Chassin et al., Reference Chassin, Pitts and Prost2002; Colder et al., Reference Colder, Campbell, Ruel, Richardson and Flay2002; Dubow et al., Reference Dubow, Boxer and Huesmann2008; Ellickson et al., Reference Ellickson, Martino and Collins2004; Englund et al., Reference Englund, Egeland, Oliva and Collins2008; Jackson & Sher, Reference Jackson and Sher2006; Li et al., Reference Li, Duncan and Hops2001; Maggs et al., Reference Maggs, Patrick and Feinstein2008; Manzardo et al., Reference Manzardo, Penick, Knop, Nickel, Hall, Jensen and Gabrielli2005; Pitkanen et al., Reference Pitkanen, Kokko, Lyyra and Pulkkinen2008; Wiesner et al., Reference Wiesner, Weichold and Silbereisen2007; Windle et al., Reference Windle, Mun and Windle2005), which includes personality (McGue et al., Reference McGue, Slutske and Iacono1999), externalizing behaviors (Boyle et al., Reference Boyle, Offord, Racine, Szatmari, Fleming and Links1992; Fergusson & Lynskey, Reference Fergusson and Lynskey1998; Helzer et al., Reference Helzer, Bucholz, Robins, Helzer and Canino1992; Lynskey & Fergusson, Reference Lynskey and Fergusson1995; Szobot & Bukstein, Reference Szobot and Bukstein2008; Young et al., Reference Young, Mikulich, Goodwin, Hardy, Martin, Zoccolillo and Crowley1995), and environmental factors such as parental monitoring (PM) (Dishion & Loeber, Reference Dishion and Loeber1985; Gillespie, Lubke et al., Reference Gillespie, Lubke, Gardner, Neale and Kendler2012), childhood sexual or physical abuse (Glaser et al., Reference Glaser, Karic and Cohen1977; Kendler, Bulik et al., Reference Kendler, Bulik, Silberg, Hettema, Myers and Prescott2000), parental attitudes toward drug use (Gillespie, Lubke et al., Reference Gillespie, Lubke, Gardner, Neale and Kendler2012; McDermott, Reference McDermott1984), household drug use (Gfroerer, Reference Gfroerer1987), deviant peer group affiliation (Jones et al., Reference Jones, Ogilvie and Ackers1976), drug availability (Freisthler, Gruenewald, Johnson et al., Reference Freisthler, Gruenewald, Johnson, Treno and Lascala2005; Freisthler, Needell, & Gruenewald et al., Reference Freisthler, Needell and Gruenewald2005; Gillespie et al., Reference Gillespie, Neale and Kendler2009), and participation in prosocial activities (Kendler & Myers, Reference Kendler and Myers2009). Among these risks, PM has been shown to be one of the most salient (Gillespie, Lubke et al., Reference Gillespie, Lubke, Gardner, Neale and Kendler2012; Latendresse et al., Reference Latendresse, Rose, Viken, Pulkkinen, Kaprio and Dick2008; Reference Latendresse, Rose, Viken, Pulkkinen, Kaprio and Dick2009).
PM is typically measured in terms of parental knowledge of children's whereabouts and degree of parental supervision. Although PM is a significant predictor of both cannabis initiation and average cannabis consumption over time (Gillespie, Lubke et al., Reference Gillespie, Lubke, Gardner, Neale and Kendler2012), the genetic and environmental association between PM and the risk of DI remains unclear. In addition to the association being driven by correlated genetic and environmental risks, DI may be phenotypically contingent upon (reduced) PM or vice versa. In lieu of genetically informative longitudinal data, alternative and innovative statistical methods can be applied to provide some capacity to infer causation. These include propensity analyses (Kendler & Gardner, Reference Kendler and Gardner2010), cross-panel designs, or modeling direction of causation based on pairs of genetically informative relatives measured on a single occasion (Heath et al., Reference Heath, Kessler, Neale, Hewitt, Eaves and Kendler1993).
Direction of causation modeling based on cross-sectional and genetically informative data has been applied to a number of complex behavioral phenotypes (Duffy & Martin, Reference Duffy and Martin1994; Gillespie, Gehrman et al., Reference Gillespie, Gehrman, Byrne, Kendler, Heath and Martin2012; Gillespie et al., Reference Gillespie, Gehrman, Byrne, Kendler, Heath and Martin2012; Neale, Walters et al., Reference Neale, Walters, Health, Kessler, Perusse, Eaves and Kendler1994). Provided several assumptions are satisfied, differences in the patterns of cross-twin, cross-trait correlation can allow researchers to falsify a hypothesis about the direction of causation between two variables measured on a single occasion (Heath et al., Reference Heath, Kessler, Neale, Hewitt, Eaves and Kendler1993). Therefore, using a genetically informative sample of male twins, our aim was to determine the direction of causation between PM and illicit DI. We fitted a model that predicted that the association between PM and the liability to DI was explained by shared genetic and environmental risk factors, which we then compared to models that predicted a reciprocal causation between PM and DI, and unidirectional causation (PM-to-DI and DI-to-PM).
Material and Methods
Subjects
Described in detail elsewhere (Kendler & Prescott, Reference Kendler and Prescott2006), this report is based on data collected from a second and third wave of interviews between 1994 and 2004 as part of an ongoing study of adult male twins from the Virginia Adult Twin Study of Psychiatric and Substance Use Disorders (VATSPUD). Briefly, twins were eligible for participation in this study if one or both twins were successfully matched to birth records, were a member of a multiple birth with at least one male, were Caucasian, and were born between 1940 and 1974. Of 9,417 eligible individuals for the first wave (1993–1996), 6,814 (72.4%) completed the initial interviews. At least 1 year later, we contacted those who had completed the initial interview to schedule the second interview. The second interview was completed by 83% of those eligible or 4,203 males comprising 1,189 complete twin pairs and 1,825 singletons. Twin pairs (no singletons) who completed the second interview were eligible to participate in a third interview (2000–2004). The VATSPUD was designed to study the nature and pattern of risk and protective factors for drug use and DUDs across adolescence and young adulthood. This third interview was completed by 75% of those eligible or 1,778 males, aged 24–62 years (μ = 40.3, SD = 9.0) from 745 complete twin pairs and 288 singletons.
In both interviews, most subjects (~90%) were interviewed by telephone. A small number were interviewed in person because of subject preference, residence in an institutional setting (usually jail), or not having a telephone. Subjects were informed about the goals of the study and provided informed consent before interviews. Interviewers had a master's degree in a mental health-related field or a bachelor's degree in this area plus 2 years of clinical experience. The two members of a twin pair were each interviewed by different interviewers. The VATSPUD was approved by the Virginia Commonwealth University institutional review board.
Zygosity and Interview Protocol
Zygosity was diagnosed using a combination of self-report measures, photographs, and DNA analysis.
Measures of Drug Initiation and Parental Monitoring
The second interview assessed lifetime drug use for six substances: cannabis (marijuana and hashish); sedatives (quaalude, Seconal, and Valium); stimulants (speed, ecstasy, and Ritalin); cocaine (intranasal, freebase, and crack); opiates (heroin, Demerol [meperidine hydrochloride], and morphine); and hallucinogens (lysergic acid diethylamide, mescaline, and phencyclidine). For substances that could be obtained legally, non-medical use was defined as use: (1) without a doctor's prescription; (2) in greater amounts or more often than prescribed; or (3) for any other reason than a doctor said it should be taken. Subjects who endorsed having ‘ever tried [drug]’ were then asked two questions: ‘How old were you the first time you took [drug]?’ and ‘How old were you when you used [drug] the most?’. A DI score (Yes/No) was then calculated for each substance and scored positive if DI was confirmed by responses to both these items. Because of the need to maintain computational efficiency, we analyzed cannabis, stimulants, and cocaine as these were the three most commonly initiated illicit substances.
Because the data collected at Wave 3 interview were retrospective, they are potentially contaminated by recall bias and telescoping effects (Pickles et al., Reference Pickles, Neale, Simonoff, Rutter, Hewitt, Meyer and Eaves1994). Therefore, our study used a Life History Calendar format (Freedman et al., Reference Freedman, Thornton, Camburn, Alwin and Young-Demarco1988) to improve the data quality. This method has been shown empirically to improve the accuracy of retrospective reporting by providing multiple cues to improve the chance of accurate recall (Belli, Reference Belli1998; Freedman et al., Reference Freedman, Thornton, Camburn, Alwin and Young-Demarco1988). This makes the task more akin to the accurate and well-retained process of recognition than to the less reliable task of free recall.
The third interview included a retrospective assessment of PM between ages 8–17 years using 11 items selected on the basis of previous work examining parental effects on risk of drug use and delinquency (Celdran et al., Reference Celdran, Bekett and Roberts1976; Ingram, Reference Ingram1976; Stattin & Kerr, Reference Stattin and Kerr2000). These items asked subjects questions such as how much their parents or guardians knew who their friends were, how they spent their money, and what they did with their free time, and where they were at night and so forth. Response options were [their parents] ‘didn't know’, ‘knew a little’, or ‘knew a lot’. PM latent factor scores were estimated for each individual in the Classic Mx software program (Neale, Reference Neale1999). Again, in order to maintain computational efficiency, we restricted our analyses to the three most prevalent PM items: ‘How much did your parents know about your friends?’; ‘How much did your parents know about how you spent your money?’; and ‘How much did your parents know about how you spent your free time?’
There were 2,576 subjects with complete DI, and 1,758 subjects with complete PM data. Depending on the substance, this included 1,749 to 1,754 subjects with both DI and PM data. For each of the three drug classes, there were 694 monozygotic (MZ) and 484 dizygotic (DZ) twin pairs, as well as 220 singletons with complete DI data. For PM, there were 454 MZ and 280 DZ twin pairs, respectively with complete data, plus 290 singletons.
Raw Ordinal Data Analysis
All data were analyzed using raw data methods in the OpenMx software implemented in R (Boker et al., Reference Boker, Neale, Maes, Wilde, Spiegel, Brick and Fox2015). This approach assumes that the observed ordinal categories within each item are an imprecise measure of a latent normal distribution of liability, and that the liability distribution for each variable has one or more thresholds that discriminate between the categories. Thresholds can be conceived of as cut points along a normal distribution that classifies individuals in terms of a probability or risk of endorsing one of two or more discrete (ordinal) categories. The DI and PM thresholds were adjusted for the linear effects of age, and before fitting univariate and multivariate models, were used likelihood ratio chi-square tests to confirm the equality of thresholds within twin pairs and across zygosity. These preliminary tests of threshold homogeneity are analogous to tests of mean and variance homogeneity in the case of continuous data.
Univariate and Multivariate Genetic Analyses
Based on standard biometrical genetic model-fitting methods (Neale & Cardon, Reference Neale and Cardon1992) that exploit the expected genetic and environmental correlations for MZ and DZ twin pairs, our models assumed that the total variance in each of the observed items and latent constructs can be decomposed into additive (A) genetic, shared environment (C), and non-shared or unique (E) environmental variance components. Because MZ twin pairs are genetically identical, correlations for the A effects are 1.0. For DZ twin pairs who on average share half of their genes, the correlations for the A effects are 0.5. An important assumption of biometrical genetic model is that shared environmental effects (C) are perfectly correlated among MZ and DZ twin pairs alike. Non-shared environmental effects are by definition uncorrelated and also reflect measurement error, including short-term fluctuations.
We used this method first to estimate the contribution of genetic and environmental risks (A, C, and E) in the univariate analyses of the PM and DI items. In the multivariate analyses, we then modeled the direction of causation between the latent factors for PM and DI because measurement error reduces the statistical power for resolving alternative causal hypotheses (Neale, Duffy et al., Reference Neale, Duffy and Martin1994; Neale, Walters et al., Reference Neale, Walters, Health, Kessler, Perusse, Eaves and Kendler1994). This approach assumes that measurement error occurs at the level of the items or indicator variables and so is uncorrelated across factors (Heath et al., Reference Heath, Kessler, Neale, Hewitt, Eaves and Kendler1993). Specifically, we compared the fit of a correlated liability model against four causal models using maximum likelihood estimation in the R-based OpenMx software package (Boker et al., Reference Boker, Neale, Maes, Wilde, Spiegel, Brick and Fox2015). The models were: (1) a comparison or correlated liability model illustrated in Figure 1a; (2) a reciprocal causation model illustrated in Figure 1b; (3–4) two unidirectional causation models. The correlated liability model is identical to bivariate Cholesky decomposition (Neale & Cardon, Reference Neale and Cardon1992) and is the null hypothesis because it predicts no causal association between the latent PM and DI factors. Instead, any phenotypic association is attributable to common or correlated genetic and environmental latent effects, that is, via the a11 and a21, c11 and c21, and e11 and e21 pathways. Under the reciprocal causation and unidirectional models, any association between PM and DI arises because of direct phenotypic causality between the latent PM and DI factors via the β12 and β21 pathways.
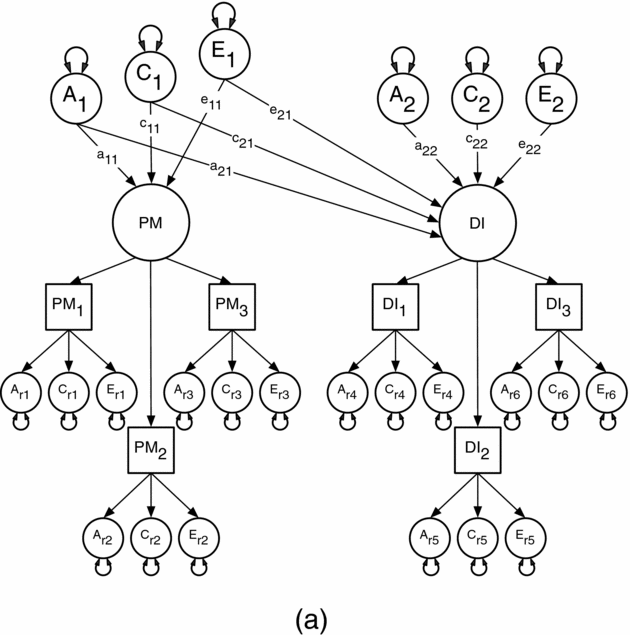
FIGURE 1a Cholesky decomposition illustrating the association between Parental Monitoring (PM) and Drug Initiation (DI) in which the sources of covariance between PM and DI are decomposed into shared genetic (a2,1) and environmental (c2,1 and e2,1) effects. This model also models the genetic (a2,2) and environmental (c2,2 and e2,2) effects unique to DI (a2,2, c2,2, e2,2).
Note: A1, C1 and E1 = latent additive genetic, shared, and non-shared environmental risks for PM, A2, C2 and E2 = latent additive genetic, shared, and non-shared environmental risks for DI, PM and DI = common factors as indicated by observed phenotypic symptoms PM1-3 and DI1-3 respectively, each with their own item specific latent genetic and environmental risk factors, PM1 = parent's knowledge of child's friends, PM2 = parent's knowledge of how child spent their money, PM3 = parent's knowledge of how child spent their free time, DI1 = cannabis initiation, DI2 = stimulant initiation, DI3 = cocaine initiation.

FIGURE 1b Illustrative model of the direction of causation between the latent factors for Parental Monitoring (PM) and Drug Initiation (DI). This model approach predicts the relationship between PM and DI is explained by a reciprocal interaction, or causal parameters (β21 and β12), at the latent factor level.
Note: A1, C1 and E1 = latent additive genetic, shared, and non-shared environmental risks for PM, A2, C2 and E2 = latent additive genetic, shared, and non-shared environmental risks for DI, PM and DI = common factors as indicated by observed phenotypic symptoms PM1-3 and DI1-3 respectively, each with their own item specific latent genetic and environmental risk factors, β21 = causal pathway from PM to DI, β12 = causal pathway from DI to PM, PM1 = parent's knowledge of child's friends, PM2 = parent's knowledge of how child spent their money, PM3 = parent's knowledge of how child spent their free time, DI1 = cannabis initiation, DI2 = stimulant initiation, DI3 = cocaine initiation.
Model Comparisons
The correlated liability model was compared to the reciprocal and unidirectional causal models using likelihood-ratio chi-squared tests with degrees in freedom being equal to the degrees of freedom between the models. The best-fitting model was chosen on the basis of parsimony, that is, the model with non-significant changes in the chi-square and the smallest number of parameters. To this end, the Akaike Information Criterion (AIC) was calculated for each model and the model with the lowest index value was chosen as the best fitting.
Results
Tests of Threshold Homogeneity and Polychoric Correlations
We found no significant differences in the threshold distribution for the three PM and three DI items within twin pairs or across zygosity. As shown in Table 1, the polychoric correlations between the DI and PM items were all moderate, ranging from -0.22 to -0.34, such that lower levels of PM were associated with increased risk of DI. The patterns of MZ and DZ twin pair correlations suggest a pattern of familial aggregation for cannabis initiation attributable to a combination of additive genetic and shared environmental risk factors. For stimulants and cocaine, the twin pair correlations suggest that familial aggregation is more likely explained by additive genetic risks alone. For the three PM items, all DZ twin pair correlations were greater than half the MZ twin pair, suggesting a combination of genetic and shared environmental risk factors.
TABLE 1 Measures of Association (Polychoric Correlations) Between Parental Monitoring and Drug Initiation Items Along With Twin Pair (Polychoric) Correlations and 95% Confidence Intervals for Each Item
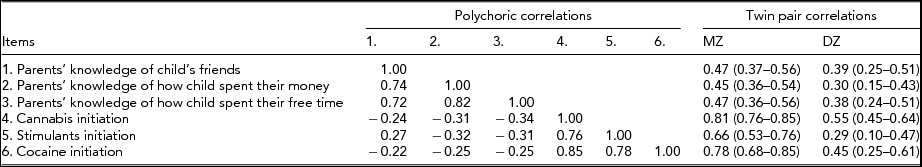
All correlations adjusted for the effects of age at interview, sex, and cohort on item thresholds.
Univariate Analyses
Standardized univariate components of variance for PM and DI items are shown in Table 2. Additive genetic risks explained between 17% and 31% of the variance in PM. Although shared environmental risk factors explained 14% to 30% of the variance in the PM items, the 95% confidence intervals spanned zero for all three PM items. Additive genetic risks explained between 42% and 64% of the variance in the DI items. Shared environmental risk factors explained 33% of the variance in the cannabis initiation. Confidence intervals for the shared environmental risks for stimulant and cocaine initiation spanned zero.
TABLE 2 Variance Components (Standardized) in the Parental Monitoring and Drug Initiation Items Attributed to Additive Genetic (A), Shared Environment (C), and Non-Shared Environmental (E) Effects Along With Their 95% Confidence Intervals

Multivariate Analysis
Results for the multivariate modeling fitting are shown in Table 3. The two unidirectional and the reciprocal causation models deteriorated significantly when compared to the (null) correlated liability model. Based on these results and the lowest AIC, the correlated liability was therefore chosen as the best fitting and most parsimonious. Standardized path coefficients for the correlated liability model appear in Figure 2.
TABLE 3 Multivariate Model Fitting Results for the (1) Non-Causal Correlated Liability, (2) Reciprocal Causation (PM↔DI), (3) Uni-Directional Parental Monitoring Causes Drug Initiation (PM→DI), and (4) Uni-Directional Drug Initiation Causes Parental Monitoring (DI→PM) Models

All PM and DI thresholds are adjusted for the linear effects of age at interview. -2LL = log-likelihood, Δ−2LL = change in log-likelihood which is asymptotically distributed as a chi-square; AIC = Akaike Information Criteria; *** p < .001.
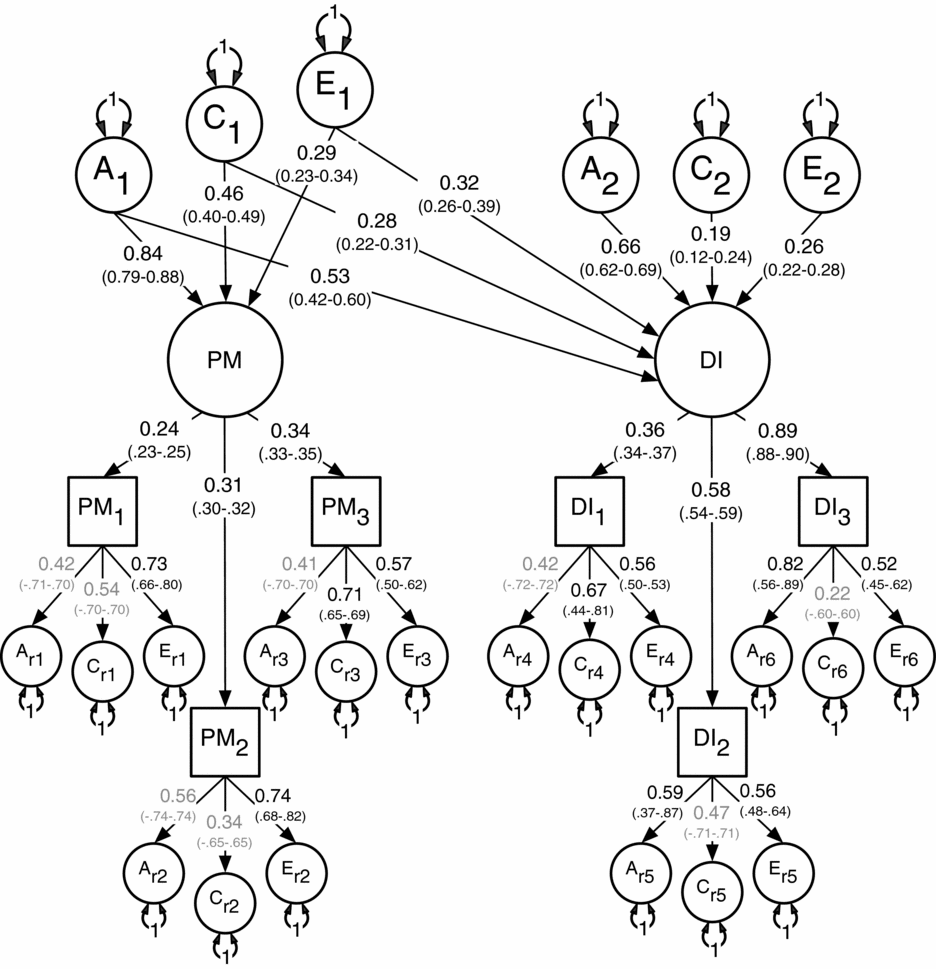
FIGURE 2 Best-fitting non-causal correlated liability model between the latent factors for Parental Monitoring (PM) and Drug Initiation (DI) with standardized path coefficients and 95% confidence intervals.
Note: PM1 = parent's knowledge of child's friends, PM2 = parent's knowledge of how child spent their money, PM3 = parent's knowledge of how child spent their free time, DI1 = cannabis initiation, DI2 = stimulant initiation, DI3 = cocaine initiation.
Discussion
PM has been assumed to be an environmental, causal risk factor for DI. Although we found a significant phenotypic association between PM and measures of DI, our data revealed that the liability to DI was not consistent with any causal hypothesis. Instead, a correlated liability model in which PM and risk of DI share common genetic and environmental risks provided a better fit to the cannabis, stimulants, and cocaine data.
Our univariate point estimates for the genetic and environmental risks in drug use were comparable to those reported elsewhere (Gillespie et al., Reference Gillespie, Lichtman, Kendler and Miller2013; Kendler et al., Reference Kendler, Chen, Dick, Maes, Gillespie, Neale and Riley2012; Verweij et al., Reference Verweij, Zietsch, Lynskey, Medland, Neale, Martin and Vink2010). Our analyses also showed that familial aggregation in PM could be explained by a combination of additive genetic effects and environmental risks. This is consistent with previous analyses based on phenotypic correlates of PM such as Parker's Parental Bonding Instrument (PBI; Parker et al., Reference Parker, Tupling and Brown1979) that have found that, depending on the informant, parenting behaviors are best explained by a combination of genetic and environmental risk factors (Gillespie et al., Reference Gillespie, Zhu, Neale, Heath and Martin2003; Kendler, Reference Kendler1996; Kendler et al., Reference Kendler, Sham and MacLean1997).
Previous reports have shown how family involvement is associated with levels of drug use even after controlling for peer influences (Chassin et al., Reference Chassin, Pillow, Curran, Molina and Barrera1993). In genetically informative studies, PM has been shown to attenuate the impact of genetic risks on other substances such as alcohol (Dick et al., Reference Dick, Latendresse, Lansford, Budde, Goate, Dodge and Bates2009; Dick et al., Reference Dick, Meyers, Latendresse, Creemers, Lansford, Pettit and Huizink2011) and nicotine consumption (Dick et al., Reference Dick, Viken, Purcell, Kaprio, Pulkkinen and Rose2007). And although the genetic risks for alcohol and nicotine use appear to be maximized under conditions of low social constraint (Dick et al., Reference Dick, Meyers, Latendresse, Creemers, Lansford, Pettit and Huizink2011; Latendresse et al., Reference Latendresse, Rose, Viken, Pulkkinen, Kaprio and Dick2008; Reference Latendresse, Rose, Viken, Pulkkinen, Kaprio and Dick2009), we found no causal impact of the PM on DI or DI on PM. Instead, covariation between self-reports of PM and the liability to DI appears to be attributable to shared, non-causal genetic, and environmental risk factors.
The idea of correlated genetic and environmental risks indexing both PM and DI is consistent with Scarr and McCartney's active model that predicts that individuals seek out environments that they find compatible and stimulating based on motivational, personality, and intellectual aspects of their genotype (Scarr & McCartney, Reference Scarr and McCartney1983). Our results are also consistent with genotype-environment correlations (CorGE), which describe situations in which an individual's environment is unlikely to be entirely random, but partly caused or correlated with his or her genotype (Scarr & McCartney, Reference Scarr and McCartney1983). Active CorGE describe cases whereby individuals ‘select’ or ‘create’ environments that are a function of their genotypes, whereas evocative CorGE describe cases whereby an individual's heritable behavior ‘evokes’ an environmental response (Neale & Cardon, Reference Neale and Cardon1992). As an example of the latter, adoptees at high genetic risk for antisocial behavior have been found to be more likely to elicit more negative parenting from their adoptive parents (O'Connor et al., Reference O'Connor, Deater-Deckard, Fulker, Rutter and Plomin1998). Both active and evocative CorGE are analogous to the unidirectional ‘drug use causes PM’ hypothesis that our data do not support. If parental knowledge derives primarily from voluntary disclosures rather than active parental surveillance (Stattin & Kerr, Reference Stattin and Kerr2000), then our results suggest that adolescents’ tendency to avoid telling their parents about their activities and whereabouts when they are engaged in drug use, or other forms of delinquent behavior, may be a function of their genetic predisposition to drug use. Another form of CorGE arises because environments in which individuals develop are provided by their biological relatives and are known as instances of passive CorGE (Neale & Cardon, Reference Neale and Cardon1992). For example, children who are genetically predisposed to drug use may also experience a pathogenic environment whereby the tendency of parents to not monitor their children may be attributable the genes also increasing the risk of DI. Therefore, a high genetic predisposition to DI is correlated with exposure to an adverse environment — for example, low PM — because both genes and environment originally derive from the parents (Neale & Cardon, Reference Neale and Cardon1992). This is consistent with our best-fitting correlated liability results. Children who are genetically predisposed to DI may live in a drug tolerant home wherein the behavioral tendency of parents to be less attentive of their children's whereabouts are underpinned by the same genes. As an example of the latter, adoptees at high genetic risk for antisocial behavior have been found to be more likely to elicit more negative parenting from their adoptive parents (O'Connor et al., Reference O'Connor, Deater-Deckard, Fulker, Rutter and Plomin1998).
Limitations
Our findings must be interpreted in the context of five potential limitations. First is the representativeness of our twin data drawn from Caucasian Virginian males. Nevertheless, our results are likely generalizable to the population at large. Although males have a higher prevalence of drug use (Kendler et al., Reference Kendler, Neale, Thornton, Aggen, Gilman and Kessler2002), previous analyses using the same data suggest that it is broadly representative of U.S. males and does not differ from the general population in rates of psychopathology, drug use, and abuse (Kendler, Karkowski et al., Reference Kendler, Karkowski, Neale and Prescott2000). Second, because the difference in fit between the reciprocal causation and the correlated liability model was marginal, the power to rule out this competing hypothesis may be insufficient. Third, our modeling was not exhaustive, and since the confidence intervals on the item-specific shared environmental factors spanned zero, these parameters were dropped in order to identify a model that simultaneously fitted both the correlated liability and the reciprocal parameters to the data. This hybrid model provided a very poor fit to the data when compared to the correlated liability model (Δ-2LL = -1.93, Δdf = 2, p < .001). Fourth, because we relied on self-reports of PM, it is unclear if drug use was associated with actual versus perceived changes in PM. Finally, demands on computational efficiency imposed by raw ordinal data analyses meant that our analyses were necessarily restricted to three PM and three DI items. We analyzed 10 additional three-item combinations from the 11 PM as well as three-item combinations of the illicit substance initiation items. In all cases, we found irrespective of which combination of PM and DI items were used, that the non-causal, correlated liability model provided the best fit to the data.
Acknowledgments
Funding was received from the US National Institute on Drug Abuse (R00DA023549 & DA-018673). This work was also supported by NIH grants AA07535 and AA07728. We also thank the twins for their cooperation.
Conflict of Interest
None.






