


Membrane protein overview
Membrane proteins are among the most fascinating structures in biology. They are by definition sited at the interface between two compartments, such as between cytoplasm and extracellular space, or between mitochondrial matrix and intermembrane space, or else they make up most of the mass of the small vesicles involved in endocytosis, exocytosis or intracellular trafficking. For most of their life, they interact closely with both water and lipid in their environment, yet must be synthesised by the ribosome just like other proteins and then make their way to different membrane locations within a cell. This places unique and sometimes conflicting demands on membrane proteins for folding, translocation and stability. Most membrane proteins function in transport or signalling or provide the structural framework that shapes cellular compartments. In signalling, they provide both the sensory input and the output, usually by involvement directly or indirectly in the release of signalling molecules. Other membrane proteins are key components of energy transduction, converting chemical energy into electrical energy, or electrical energy into either mechanical energy or synthesis of ATP, the universal energy currency of the cell.
Knowledge of their structure tells us how they are oriented relative to the lipid bilayer and often suggests how they work. As a result, the structure of membrane proteins provides a rich source of information in biology. In the very practical search for better drugs to improve human and animal health, many targets are membrane proteins involved in signalling or growth control at the cell surface. Over the last 20 years, there has been enormous progress in understanding membrane protein structure (Fig. 1). With over 150 unique structures now available and multiple sets of coordinates deposited for many structures in the Protein Data Bank (PDB), it is an excellent moment to review the field.
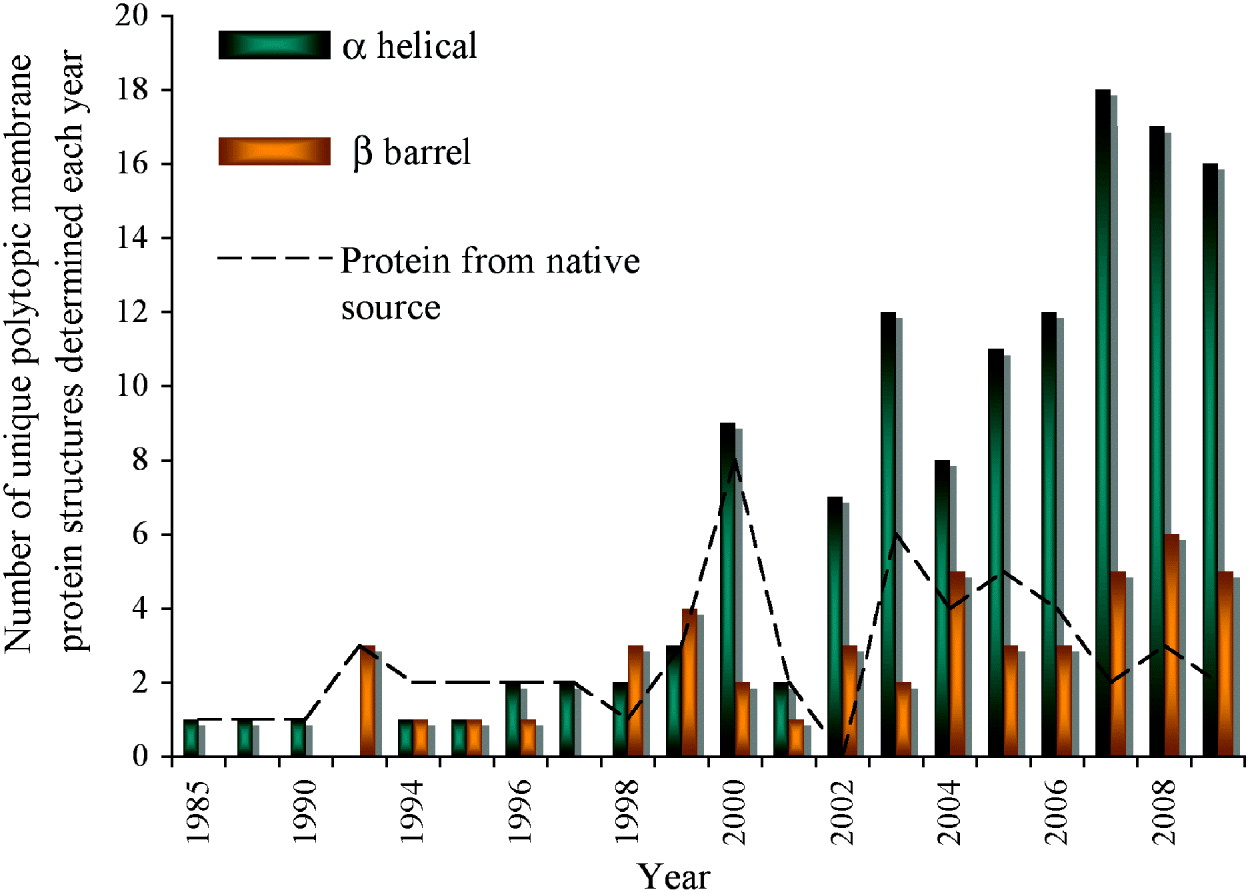
Fig. 1. Progress of membrane protein structure determination. Starting with the first structure in 1985, 174 unique membrane protein structures have been determined till the end of 2009. However, the Protein Data Bank (PDB) holds many more than this with for example, over 60 coordinates each for reaction centres and bacteriorhodopsin alone. We have included in the chart only polytopic membrane proteins that have a functional role within the membrane and not intrinsic membrane proteins with only a single, presumably regular trans-membrane α-helix. Mutants, different conformational states, structures with bound substrates/inhibitors of the same protein, or membrane proteins from different species with >70% sequence homology are counted only once. There are numerous ways of classifying membrane protein structures: here we present the distribution classified on the basis of α-helical or β-barrel secondary structure; a different classification on the basis of prokaryotic or eukaryotic origin can be found elsewhere (Carpenter et al. Reference Carpenter, Beis, Cameron and Iwata2008). In the early years, structures were determined from proteins that were abundant in their natural environment including the reaction centres (1985 and 1987), bacteriorhodopsin (1990), porins (1992), light harvesting complex (1994) followed by a variety of electron transport and photosynthesis complexes. The first structures of membrane proteins expressed recombinantly started to emerge from 1998 (KcsA, MscL, OmpA and FhuA). Since then, the availability of sequenced genomes in the late 1990s propelled the rate of membrane protein structure determination, which has reached its highest level in the past two years. The following link provides a complete list of available structures with links to the PDB (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html). In this review, we give the PDB accession number for the structures shown in the figures.
Historically, it was not until the invention of SDS-polyacrylamide gel electrophoresis (Shapiro et al. Reference Shapiro, Vinuela and Maizel1967; Weber & Osborn, Reference Weber and Osborn1969; Laemmli, Reference Laemmli1970) and its early application to the membranes of red blood cells (Lenard, Reference Lenard1970) and rod outer segments (Heitzmann, Reference Heitzmann1972) that our understanding of membrane structure progressed from the unit membrane hypothesis of Danielli & Davson (Reference Danielli and Davson1935) and Robertson (Reference Robertson1957) to the fluid mosaic model (Singer & Nicolson, Reference Singer and Nicolson1972). Before the advent of recombinant DNA technologies, research efforts were limited to membrane proteins available in reasonable quantities from natural sources. This period represented the first phase of membrane protein structure determination. It included work on membrane proteins from chloroplasts, mitochondria and bacteria that were involved in energy transduction or other functions that require high levels of expression. These high expression levels in native cells were usually accompanied by low turnover rates, and this correlated with relatively good stability in the fairly restricted range of detergents available initially. More powerful cloning methods involving novel leader sequence or whole protein fusions to increase expression levels, better detergents and the invention of tags such as polyhistidine (Hochuli et al. Reference Hochuli, Bannwarth, Dobeli, Gentz and Stuber1988) that allowed rapid purification have now made it possible to express and purify any membrane protein for which the gene or cDNA had been identified. However, progress in membrane protein structure determination was still slow because many, perhaps most, membrane proteins turned out to be relatively unstable in detergent and therefore difficult to work with. The normal environment of a membrane protein in a lipid bilayer includes contact with a ring of closely packed headgroups on each side of the lipid bilayer with the most fluid, mobile part of the bilayer being in the middle of the membrane. In contrast, the detergent micelle that surrounds a solubilised membrane protein provides the opposite situation in which the least ordered, most mobile part of the micelle is in the surface region normally occupied by the lipid headgroups. In addition, some of the earliest membrane protein structures determined were those of proteins involved in photosynthesis or electron transport, in which the proteins function to provide a rigid environment for fixed cofactors. This rigidity contributed to their stability and helped to produce well-ordered crystals.
The second phase of successful membrane protein structure determination did not start until the complete sequences of bacterial genomes started to become available in the 1990s. By searching the genome sequences of a range of mesophilic, thermophilic or hyperthermophilic bacteria for homologous membrane protein genes and then screening a large number of these for good expression, stability and crystallisation, there has been considerable success in the determination of many prokaryotic membrane structures. One of the earliest examples was the work on bacterial mechanosensitive channels, MscL (Chang et al. Reference Chang, Spencer, Lee, Barclay and Rees1998). In other cases, the proteins were often of unknown biological function in the bacterium from which they were obtained. Nevertheless, their sequence homology with other membrane proteins of known function meant that the determination of their structure was a great step forward because it provided a connection of structure to function for some important membrane protein families. Early successes of this approach, which has had a substantial impact on our understanding of the architecture of membrane proteins, were members of the bacterial potassium channel family (MacKinnon, Reference Mackinnon2004b) and the ABC transporter family (Locher et al. Reference Locher, Lee and Rees2002). Many other types of bacterial channels and transporters have now joined these early successes.
In recent years, we have arguably entered a third phase of membrane protein structure analysis with the successful demonstration of generic strategies to stabilise and crystallise unstable, eukaryotic membrane proteins. Although many membrane proteins, especially those from eukaryotes, have evolved to be adequately stable in vivo in lipid bilayers, which are often made less fluid by the presence of cholesterol or other rigidifying membrane components, they can be highly unstable in the detergent micelles into which they must be extracted for purification, and even less stable in some of the smaller, harsher detergents that have been most successful in crystallisation. Indeed, the rapid synthesis, turnover and degradation of a typical membrane protein is often important for its normal dynamic function, allowing a rapid response to different demands on the cell. During evolution, proteins are subjected to many pressures, so that they tend to be only as stable as they need to be in their normal lipid environment. For crystallisation, such unstable membrane proteins must be stabilised either by timely reinsertion into a lipid bilayer, such as with 2D membrane crystals (Kuhlbrandt, Reference Kuhlbrandt1992) or with lipidic phase, detergent-free crystallisation methods (Nollert et al. Reference Nollert, Royant, Pebay-Peyroula and Landau1999), or by the addition of specific active-site ligands or inhibitors (Pebay-Peyroula et al. Reference Pebay-Peyroula, Dahout-Gonzalez, Kahn, Trezeguet, Lauquin and Brandolin2003; Vedadi et al. Reference Vedadi, Niesen, Allali-Hassani, Fedorov, Finerty, Wasnet, Yeung, Arrowsmith, Ball, Berglund, Hui, Marsden, Nordlund, Sundstrom, Weigelt and Edwards2006; Toyoshima, Reference Toyoshima2008), or by systematic mutagenesis to create a protein with more intrinsic stability (Magnani et al. Reference Magnani, Shibata, Serrano-Vega and Tate2008; Serrano-Vega et al. Reference Serrano-Vega, Magnani, Shibata and Tate2008). This third post-genomic phase should allow the structure of any membrane protein or complex of interest to be determined.
Structures of membrane proteins follow simple rules governed by their hydrophobic nature and the restrictions posed by the lipid bilayer. Following the determination of the seven transmembrane (TM) helix structure of bacteriorhodopsin (Henderson, Reference Henderson1975), it was thought that TM proteins might consist of either α-helical bundles or β-barrels, because the fully satisfied backbone hydrogen-bonding found within these two classes of structure would avoid unfavourable interactions of backbone amido or carbonyl groups with the hydrophobic lipid bilayer environment (Henderson, Reference Henderson, Balian, Chabre and Devaux1981). With a few extremely informative exceptions, which will be discussed further in this review, these two types of structures have turned out to be the predominant structural theme of all membrane proteins, with α-helical bundles being found almost exclusively in cytoplasmic and subcellular compartment membranes and β-barrels being found almost exclusively in the outer membranes of bacteria, mitochondria and chloroplasts. A deviation from regular α-helix is frequently observed in membrane proteins usually to satisfy some aspect of function and will be highlighted in the review when possible. In the area of membrane protein biogenesis, the initial hypothesis of helical hairpin insertion (Engelman & Steitz, Reference Engelman and Steitz1981) was followed by the two stage model (Popot & Engelman, Reference Popot and Engelman2000), discussions of the physical principles (White & Wimley, Reference White and Wimley1999) and the positive-inside rule (von Heijne, Reference Von Heijne2006) with recent considerations of additional steps (Engelman et al. Reference Engelman, Chen, Chin, Curran, Dixon, Dupuy, Lee, Lehnert, Matthews, Reshetnyak, Senes and Popot2003) and the idea of significant protein and lipid fluidity. However, the kinetics of membrane protein synthesis, insertion and degradation will not be covered in this review, which will focus purely on structure.
Channels and pores
The term channel or pore implies an opening in the membrane through which a molecule or ion can pass that, depending on specificity, may or may not involve a binding and recognition step. Some of these proteins form selective channels conducting a particular ion, others select for cations or anions, and yet others are non-selective. Channels can be gated by voltage or a ligand. Channel-forming proteins that are ion-selective usually have pores lined with charged amino acids or electrostatic dipoles. The width of the pore determines whether an ion flows through the channel in a hydrated or dehydrated state. Non-selective channels have wider pores that allow the ions to pass in their hydrated state. The probability of ions shedding their hydration shell is greatly increased when the pore is narrow with charges and dipoles on the wall of the channel. A cation channel has negatively charged residues within or near the entrance of the pore to attract cations and repel anions while the reverse is true for anion specific channels. There are also channels that conduct small molecules such as water, glycerol, ammonia and cAMP. Although these small molecule channels have a different architecture they share some of the properties of ion channels. We discuss below the known structures of channels classified by their type and function.
Tetrameric ion channels
There has been a great deal of progress in understanding ion channels in membranes, particularly voltage-gated ion channels. The overall architecture of this family shows a tetrameric arrangement of identical subunits or a single polypeptide linking together four homologous repeats, or in rare intermediate cases two polypeptides with two repeats each. The channels fall into two further classes: simpler channels have two TM α-helices per subunit whereas more complex channels have six TM α-helices. From the point of view of membrane protein structure, a number of interesting principles are revealed by these structures. These include the nature of the ion pores, how they open and close, and how the TM electric field can be sensed and coupled to channel opening and closing.
The most significant step forward in understanding the structural basis of the ion specificity and flux came from the first structure of the bacterial potassium channel (Doyle et al. Reference Doyle, Morais Cabral, Pfuetzner, Kuo, Gulbis, Cohen, Chait and Mackinnon1998) KcsA in 1998, the importance of which was acknowledged by award of the 2003 Nobel Prize for Chemistry to Rod MacKinnon (MacKinnon, Reference Mackinnon2004a), shared with Peter Agre for his work on water channels, which are presented in section 2.7. The original KcsA structure consisted of 97 amino acids in each of the four subunits. It showed the inner of the two TM helices of each subunit forming an inverted teepee centred on the molecular fourfold axis.
The short re-entrant loop between the two TM helices, consisting of a ten-residue pore helix and a four-residue stretch of β-structure (β-pore) in each subunit that forms the selectivity filter, is located within the teepee (Fig. 2a). The carboxy termini of the four pore helices point directly at a central cavity at the core of the protein, and the carbonyl groups of the short selectivity filter line the narrowest part of the pore near the extracellular surface. The dipoles of these short helices may help to stabilise the cation when it is at the centre of the membrane and the carbonyl groups provide the lining for the pore of the K+-ion selectivity filter (Doyle et al. Reference Doyle, Morais Cabral, Pfuetzner, Kuo, Gulbis, Cohen, Chait and Mackinnon1998). The tight turn between the pore helix and the β-pore partly lines the central aqueous cavity. Subsequent higher resolution structures, in complex with an Fab antibody fragment, showed the potassium ion coordination and hydration in more detail and how the structure adapts to low and high potassium concentration (Zhou et al. Reference Zhou, Morais-Cabral, Kaufman and Mackinnon2001) as the channel opens and is exposed to the higher intracellular potassium concentration. The central cavity accommodates a single highly hydrated potassium ion surrounded by eight ordered water molecules, plus additional partially ordered and disordered water molecules. The narrow selectivity channel can accommodate a line of potassium ions interspersed with water molecules. The role of the selectivity filter can be appreciated by comparing KcsA with a non-selective cation NaK channel from Bacillus cereus (Shi et al. Reference Shi, Ye, Alam, Chen and Jiang2006).

Fig. 2. Tetrameric potassium channels: (a) KcsA (1K4C) and (b) Kv1.2/Kv2.1 chimera (2R9R) with extracellular side at the top. Potassium ions are shown as red spheres. The prokaryotic channel KcsA represents the simplest potassium channel with two TM helices. Each subunit is coloured individually. The extracellular (top) section of the pore has a stretch of β-structure conserved by evolution for potassium ion selectivity. The gating of KcsA is thought to occur by a change in pH, with the channel opening at acidic pH (Heginbotham et al. Reference Heginbotham, Lemasurier, Kolmakova-Partensky and Miller1999). Many other K+ channels have a more complex architecture with six TM helices. The structure of the Kv1.2/Kv2.1chimera (Long et al. Reference Long, Tao, Campbell and Mackinnon2007) is one such example. The pore forming helix S5 and outer helix S6 are coloured as in KcsA. The voltage sensing S4 helix (dark blue) is replete with arginines that are thought to move in response to a change in membrane potential resulting in opening or closing of the channel. Voltage-gated K+ channels also have a β-subunit (light orange) that is essential for regulation and makes contact with the TM domain via linker T1 (brown). The structure of KcsA depicts a closed channel while the Kv1.2/Kv2.1 chimera is probably an open depolarised state.
It has been proposed that the structure obtained for another bacterial potassium channel, MthK (Jiang et al. Reference Jiang, Lee, Chen, Cadene, Chait and Mackinnon2002), is representative of the open state of this family of channels, since its structure shows a large movement of the cytoplasmic half of the inner helix, with a pronounced 30° bend at a glycine residue to create a 12-Å wide channel opening. The high ion flow rate and selectivity of the channels can be explained by the structures (Jiang et al. Reference Jiang, Lee, Chen, Cadene, Chait and Mackinnon2002). The helix bending that underlies gating in different potassium channels may occur at this or other glycine or proline residues along the inner helix, such as in KirBac1.1 (Kuo et al. Reference Kuo, Gulbis, Antcliff, Rahman, Lowe, Zimmer, Cuthbertson, Ashcroft, Ezaki and Doyle2003).
The mechanism of the opening and closing of the six-helix voltage-dependent sodium and potassium channels has been less easy to explain than the simpler gating of the two-helix potassium-selective channels, because of their greater complexity and larger number of functional states. The six-helix channels are of great importance since their function underlies all mammalian nerve and muscle function (Tempel et al. Reference Tempel, Papazian, Schwarz, Jan and Jan1987; Catterall, Reference Catterall2000). Electrophysiological measurements and site-directed mutagenesis of both native and chimeric proteins have shown that the first four helices, named segments S1–S4, form the voltage sensor domain. This S1–S4 domain appears to be relatively independent and interacts with the tetrameric, channel forming, S5–loop–S6 domain, to open and close the channel in response to membrane potential. The structure of the S5–loop–S6 domain from six-helix potassium channels is very similar to that of two-helix KcsA channels (Jiang et al. Reference Jiang, Lee, Chen, Ruta, Cadene, Chait and Mackinnon2003a). Along S4, a series of four to seven positively charged arginine or lysine residues (named R0 to R6 in Kv2.1), spaced every three residues, has been shown to respond to the membrane potential by moving across the electrical width of the membrane.
Structures for 4 six-helix potassium channels have been determined (Jiang et al. Reference Jiang, Lee, Chen, Ruta, Cadene, Chait and Mackinnon2003a; Long et al. Reference Long, Campbell and Mackinnon2005a; Reference Long, Campbell and Mackinnon2005b, Reference Lovering, De Castro, Lim and Strynadka2007). The earliest structure KvAP (Jiang et al. Reference Jiang, Lee, Chen, Ruta, Cadene, Chait and Mackinnon2003a, Reference Jiang, Ruta, Chen, Lee and Mackinnonb) is now thought to show non-physiological conformations for S1–S4, in which the harsh detergent, octyl-glucoside, used for crystallisation has perturbed the structure. The second structure Kv1.2 (Long et al. Reference Long, Campbell and Mackinnon2005a) used milder detergents plus added lipid in crystallisation, so it shows a structure with the molecules arranged in membranous layers, and likely to represent the open form occurring in depolarised membranes where there is no membrane potential. However, this second structure was at relatively low resolution so did not allow reliable identification of side chain identity. The third structure (Long et al. Reference Long, Tao, Campbell and Mackinnon2007) of a Kv1.2/Kv2.1 chimera, also crystallised using mild detergents plus lipids, has higher resolution with clear side-chain density, a more continuous polypeptide chain and ordered interhelical loops. It is currently the best structure available representing the open, depolarised state of a six-helix voltage-dependent ion channel (Fig. 2b). It shows a compact four-helix bundle in which five of the seven positively charged residue positions in S4 are accessible on the outside surface of the membrane either directly to solvent or via hydrogen bonds to a negatively charged cluster of glutamate side chains, as expected and required for opening of the voltage-gate in the depolarised state.
The structure of a closed, hyperpolarised form of any six-helix voltage-gated channel is unknown and may be difficult to determine, since it normally requires membrane hyperpolarisation to around −100 mV, which is difficult to create in a crystal. Nevertheless, a hypothetical mechanism for voltage gating has been proposed (Long et al. Reference Long, Tao, Campbell and Mackinnon2007) that involves a major 12- to 15-Å inward sliding movement of S4 relative to S1 and S2 (to close the channel), with a smaller accompanying movement of S3b. In this proposed S4 translation, possibly accompanied by a concertina-like shift of the 310-helical region along S4, the location of the positively charged side chains in S4 move from being extracellular to being effectively intracellular. This proposed movement to explain the mechanism of voltage gating is smaller and subtler than the paddle-like movement proposed earlier (Jiang et al. Reference Jiang, Lee, Chen, Ruta, Cadene, Chait and Mackinnon2003a). Residue arginine R2, for example, hypothetically moves from being exposed on the outside surface in the open state to forming a salt bridge with the conserved glutamic acid in S2 that is effectively on the inside. The predominance of arginine rather than lysine side chains in S4 and the interaction of some of them with other negatively charged side chains may reduce the energy barrier for crossing the membrane by distributing the charge more widely, as noted previously (Jiang et al. Reference Jiang, Ruta, Chen, Lee and Mackinnon2003b). Coupling of the voltage-dependent structural rearrangement of S4 is likely to involve the exercise of a mechanical force on the S4–S5 linker which then pulls open the channel gate formed by the cytoplasmic ends of the S6 inner helices (Long et al. Reference Long, Tao, Campbell and Mackinnon2007).
Finally, the TM region of a bacterial cyclic nucleotide-regulated channel, MlotiK1 represents another class of potassium channels (Clayton et al. Reference Clayton, Altieri, Heginbotham, Unger and Morais-Cabral2008). MlotiK1 is not voltage-gated, yet has an S1–S4 domain that is a slightly more compact α-helical bundle than in Kv1.2/Kv2.1. The key positive and negatively charged side chains present in S4 of the voltage-gated channels are uncharged in MlotiK1. The opening of this channel appears to involve a direct bending of the cytoplasmic end of S6 triggered by a structural change when cyclic nucleotide binds to the cyclic nucleotide-binding domain that is covalently connected to the C-terminus of S6.
Pentameric ligand-gated ion channels
The pentameric ion channels make up a well-conserved but widespread family (Tasneem et al. Reference Tasneem, Iyer, Jakobsson and Aravind2005) that has many important roles in human physiology, the best known being that of chemical signalling at the neuromuscular junction. The channels are ligand-gated though, in some bacterial homologues, the ligand may be as small as a hydrogen ion. They form homopentameric or heteropentameric assemblies, with the nicotinic acetylcholine receptor, having a subunit composition of α2βγδ, being the most studied. The α-, β-, γ- and δ-subunits are homologous, with the two α-subunits being responsible for ligand binding. In vertebrates, members of this family include the GABAA, glycine and 5-HT3 receptors. By comparison, the equally important glutamate ion channels, including AMPA, kainate and NMDA are probably related to the tetrameric potassium voltage-gated ion channels discussed in section 2.1.
The nicotinic acetylcholine receptor (AChR) structure has been determined at 4-Å resolution by electron microscopy of helical arrays from the Torpedo electric organ (Miyazawa et al. Reference Miyazawa, Fujiyoshi and Unwin2003; Unwin, Reference Unwin2005). The interpretation of the density map was helped by a knowledge of the structure of a homologous non-membrane protein, the acetylcholine binding protein (AChBP) from mollusc synapses (Celie et al. Reference Celie, Van rossum-Fikkert, Van Dijk, Brejc, Smit and Sixma2004), which was shown to have a two sheet β-sandwich structure using X-ray crystallography. The acetylcholine receptor thus consists of a pentamer of the extracellular β-domains, which form the N-termini of the polypeptides, attached to the transmembrane C-terminal domains with four transmembrane helices per subunit, making 20 transmembrane helices altogether (Fig. 3a). The structure of AChBP in complex with acetylcholine and other ligands also showed that the ligand-binding site lies at the boundary between adjacent subunits in the pentamer. In the case of a homopentamer like AChBP, there are five binding sites per pentameric molecule, whereas the nicotinic acetylcholine receptor has two at the interfaces between the α-γ and α-δ subunits.

Fig. 3. Pentameric ligand-gated channels, side view of full-length protein and top view of TM domain only: (a) AchR (2BG9), (b) ELIC (2VL0) and (c) GLIC (3EAM). Ligand-gated channels are non-selective cation channels that form homo or hetero pentamers. Each subunit in the figure is coloured individually. In contrast the major voltage-gated channels are tetrameric (shown in Fig. 2) and are selectively permeable to K+ or Na+ ions. In higher organisms, ligand-gated channels play a major role in signalling, the best-known example being the acetylcholine receptor (AChR) in the neuromuscular junction. The recent identification of prokaryotic pentameric ligand-gated channels (ELIC and GLIC) through genomic homology searches has allowed the determination of two high-resolution X-ray structures but their physiological function remains to be elucidated. Each subunit of all channels in this family has four TM helices. A large extracellular domain binds the ligand, acetylcholine in AChR, protons in GLIC and unknown in ELIC. In AChR, the pentamer composition is α2βγδ-subunits with only the two α-subunits binding the ligand. Structures of AChR determined by EM and ELIC by X-ray crystallography reflect the closed state of channel, while GLIC crystallised at low pH is probably an open state of the channel. The outward tilting of the inner helices in GLIC, proposed as the basis of channel opening, can be seen in the top view.
Two recent X-ray structures of bacterial homologues from the same family of pentameric ligand-gated ion channels have confirmed the overall molecular architecture and provided higher resolution details. In the first case (Hilf & Dutzler, Reference Hilf and Dutzler2008), the structure of ELIC (Erwinia chrysanthemi ligand-gated ion channel) appears to represent a closed channel though the ligand that opens it is unknown (Fig. 3b). In the second case (Bocquet et al. Reference Bocquet, Nury, Baaden, Le Poupon, Changeux, Delarue and Corringer2009; Hilf & Dutzler, Reference Hilf and Dutzler2009), the structure of GLIC (Gloebacter violaceous ligand-gated ion channel) has been solved by two independent groups simultaneously and appears to show an open channel conformation (Fig. 3c). In GLIC, the ligand is believed to be a hydrogen ion that may protonate an aspartate side chain in the extracellular domain leading to channel opening. By comparing the GLIC and ELIC structures, the authors suggest that channel opening may occur by a combined tilting of both M3 and central M2 helix by 9° so that the intracellular end of M2 enlarges the channel diameter from essentially zero (closed) to about 5 Å (open).
Hexameric ion channels
In multicellular organisms, exchange of small molecules such as ions, metabolites and nucleotides between neighbouring cells can be mediated through specialised channels called gap junctions. Each gap junction complex consists of two hemichannels, called connexons, that interact end to end to form a continuous channel thereby connecting the cytoplasm of two cells and bypassing the extracellular space. Each connexon consists of six monomers of the protein connexin. Based on sequence homology, human connexins have been classified into three isoforms (α,β,γ) that give unique properties to a particular gap junction. Gating by voltage, calcium, pH or phosphorylation has been observed in gap junctions (Harris, Reference Harris2001). The physiological function of Ca2+ or H+ gating may be to protect undamaged cells from neighbouring cells that suffer damage and are dying.
Each connexin monomer has four TM helices, two extracellular loops, one cytoplasmic loop, an N-terminal helix and a C-terminal segment. The structure of human connexin 26 gap junction reveals a 38-Å thick membrane region with TM2 extending ~19 Å into the cytoplasm (Maeda et al. Reference Maeda, Nakagawa, Suga, Yamashita, Oshima, Fujiyoshi and Tsukihara2009). The extracellular region extends 23 Å from the membrane surface and interacts with a connexon from the opposing cell, resulting in an intercellular gap of ~40 Å. The resolution of the density map was not good enough by itself to allow unambiguous interpretation of the structure, so the location of selenomethionine labels was needed to build the model. Helices TM1 and TM2 face the interior of the pore while TM3 and TM4 face the exterior (Fig. 4). This condradicts an earlier hypothetical model based on a 3D map of connexin from 2D crystals plus other considerations, which placed TM1 and TM3 facing the interior of the pore (Unger et al. Reference Unger, Kumar, Gilula and Yeager1999; Fleishman et al. Reference Fleishman, Unger, Yeager and Ben-Tal2004). However, it should be emphasised that the experimental maps obtained by electron microscopy of 2D crystals and X-ray diffraction of 3D crystals are virtually identical and that some of the surface loops were unresolved in both maps, so the current structure should probably still be considered as provisional.

Fig. 4. Gap junction (2ZW3): (a) side view of complete gap junction and (b) top view (from cytoplasm) of a hemichannel. Gap junctions are made up of connexin monomers that assemble into hexameric rings called connexons. A complete structure of a gap junction consists of two apposed connexons mediated by strong interactions between the extracellular domains and a continuous open channel that connects the cytoplasm of adjacent cells. A connexin monomer [highlighted in colour in the side view (a)] has four TM helices with surface loops connecting them. TM1 (dark blue) and TM2 (light blue) form the wall of the pore. TM3 and TM4 (light orange) form the outer helices. The extracellular loops 1 and 2 (aquamarine) form most of the interactions between the two interacting connexons and seal the junction from the extracellular environment. The putative voltage sensing N-terminal helix is shown in salmon.
Like in many channels, a proline residue introduces a kink at the midpoint of TM2 and a mutation of this residue results in aberrant gating. The major pore-lining helix TM1 is tilted outwards from the pore axis, resulting in narrowing of the channel towards the extracellular side of the membrane. A prominent feature in TM3 is the occurrence of an aromatic residue every third or fourth position. Although TM3 is least conserved in connexins, aromatic residues are involved in interactions between adjacent protomers. The diameter of the pore is widest at the cytoplasmic entrance measuring 40 Å but narrows to 14 Å near the extracellular membrane surface. The width of the channel increases again in the extracellular space to 25 Å. The cytoplasmic entrance formed mainly by TM2 and TM3 exposes many positively charged residues that should concentrate negatively charged molecules. The N-terminal helix forms a constriction on the cytoplasmic side and may play a role in selectivity. In the present structure, residues from the N-terminal helix interact with residues from neighbouring monomers and probably help to maintain the channel in an open state.
The extracellular loops E1, E2 and the extracellular halves of TM2 and TM4 mediate the interaction between two connexons. A most important requirement of this intercellular junction is to prevent leakage of any cellular material from intracellular to extracellular compartments. E1 contains a 310 helix at its N-terminus and a short helix at its C-terminus while E2 has a flexible N-terminus and a C-terminal half with a 310 turn. The N-terminal half of loop E1 forms the inner wall of an extracellular cavity. β-strands from E1 and E2 form an antiparallel β-sheet that covers E1 and the extracellular cavity, thereby forming an outer wall. There are a number of interactions involved between the E1 and E2 loops from one hemichannel and the same loops from the opposite hemichannel resulting in a tightly sealed junction. The position of the N-terminal helix and earlier electron crystallographic studies has led to the proposal that it could act as a plug (Oshima et al. Reference Oshima, Tani, Hiroaki, Fujiyoshi and Sosinsky2007).
Trimeric ion channels
Exclusively found (so far) in higher eukaryotes are cation selective, voltage-independent, ligand gated trimeric ion channels with two TM helices and a large extracellular domain inserted between them. This architecture is quite different from the tetrameric potassium channels or the pentameric ligand-gated channels. Two families of trimeric ion channels include the degenerin/epithelial sodium channel (DEG/ENaC) and the P2X receptors. The DEG/ENaC family includes the peptide-gated channels of molluscs, touch sensitive degenerins of Caenorhabditis elegans, constitutively open channels in lung and kidney epithelia involved in Na+ reabsorption, and acid sensing ion channels (ASICs) found in the nervous system. The N and C-termini of the protein are cytoplasmic and mediate protein-protein interactions (Bianchi & Driscoll, Reference Bianchi and Driscoll2002; Wemmie et al. Reference Wemmie, Price and Welsh2006); some of members of this family have been implicated in mechanotransduction. Proteins from DEG/ENaC family show higher selectivity for sodium than other cations. Proteins with similar architecture but unrelated to DEG/ENaC family are the P2X receptors found both in pre and post-synaptic neurons of the central nervous system. In post-synaptic neurons, extracellular ATP acts as a ligand to open a non-selective cation permeable channel. In afferent neurons of the peripheral nervous system, the P2X receptors are involved in sensing taste and pain. In both these families, as one would expect, the largest variations are observed in the extracellular domains, which have evolved to bind different ligands. Unlike in the other families of ion channels, lack of homologous proteins in prokaryotes meant the structure of trimeric channels had to be determined from eukaryotic sources. Two structures, chicken ASIC of the DEG/ENaC family (Jasti et al. Reference Jasti, Furukawa, Gonzales and Gouaux2007; Gonzales et al. Reference Gonzales, Kawate and Gouaux2009) and zebrafish P2X4 (Kawate et al. Reference Kawate, Michel, Birdsong and Gouaux2009) have been determined to reveal a similar architecture and some common principles.
Acid sensing ion channels
Six isoforms of ASICs (1a, 1b, 2a, 2b, 3 and 4) have been identified in mammals and are distributed both in the central and peripheral nervous system where a drop in extracellular pH can activate these channels (Krishtal, Reference Krishtal2003; Wemmie et al. Reference Wemmie, Price and Welsh2006). Two X-ray structures of ASIC1 from chicken have been determined that differ in terms of the construct used and their functional state (Jasti et al. Reference Jasti, Furukawa, Gonzales and Gouaux2007; Gonzales et al. Reference Gonzales, Kawate and Gouaux2009). The pH of half maximal activation of chicken ASCI is 6·7, and it desensitises upon prolonged exposure to low pH. The first structure of chicken ASCI lacks the first 25 and last 64 residues at the N- and C-terminus, respectively, and does not produce proton-induced currents. Since the crystals were grown at pH 5·6, it is thought to represent a closed desensitised state of a non-functional channel (Jasti et al. Reference Jasti, Furukawa, Gonzales and Gouaux2007). The construct used for the second structure includes all the residues at the N-terminus intact but lacks 61 residues at the C-terminus (Gonzales et al. Reference Gonzales, Kawate and Gouaux2009). This construct does elicit proton-induced currents and sodium selectivity. Thus, the structure, solved at pH 6·5, has been described as a desensitised state of a functional channel.
The extracellular domains of the two structures are identical and reveal interesting features. Three large β-sheets from each subunit form the core of the extracellular domain, which is then surrounded by a mixture of α and β structures. The salient feature of the extracellular domain in the DEG/ENaC family is the presence of conserved cysteine rich regions. A total of seven disulphide bonds are found. Of these, five are arranged in a straight line terminating at a conserved tryptophan residue located at the junction between the TM and extracellular domains. Two β-strands connect the TM domain to the extracellular domain and well-defined loops are found at the membrane interface. The extracellular domain is filled with many crevices and cavities that might interact with other proteins. In ASICs, the ligands are protons. An acidic pocket found 45 Å away from the membrane in the extracellular domain might act as a pH sensor. It is speculated that this cysteine rich domain conveys the proton-induced conformational change of the extracellular domain to the TM domain through loops at the membrane interface, resulting in opening or closing of the channel. The extracellular domain has vestibules with a negative potential that can act as a reservoir for cations. In the current structures there is no continuous pore along the threefold axis to the TM domain but ions are thought to access the pore through fenestrations near the membrane surface.
As expected for a cation-selective channel the interior of the TM domain has a negative potential. Residues from TM2 primarily provide the pore lining, but a few residues from TM1 line the extracellular side of the pore. A conserved Gly–Ala–Ser motif is thought to play a crucial role in ion selectivity. The TM domains in the two structures of ASCI show substantial differences (Jasti et al. Reference Jasti, Furukawa, Gonzales and Gouaux2007; Gonzales et al. Reference Gonzales, Kawate and Gouaux2009). In the first structure, the TM domains in each of the three subunits differ in their conformation. The TM2 helices of two of the subunits have a substantial kink at a glycine residue while the helix in the third subunit is relatively straight. The packing of helices results in a V-shaped structure that opens to the extracellular side (Fig. 5a). As a result of the kinks, two TM2 helices cause leucine side chains to occlude the pore and no bound ions are found in the pore, hence this structure is called a closed state of the channel. In the second structure, the TM domains of all three subunits are identical. TM1 probably interacts with the lipids that cover TM2. The inner TM2 helix is tilted ~50° and the three TM2 helices cross each other halfway from the putative membrane boundary. Such an arrangement of TM helices results in wide extracellular and cytoplasmic regions with a narrow constriction at the middle of the pore. However, the leucine residues no longer occlude the pore (Fig. 5b), but aspartates from all three helices contribute to a constriction on the extracellular side. Soaking with caesium ions reveals an elongated density at this extracellular vestibule coordinated by aspartate and glycine. Does this represent a different conformation of the channel? It should be noted that the crystal packing in the two structures is different. In the first structure, the TM domains mediate the interaction between layers of the extracellular domain: this crystal contact may fix the TM domains, which may then show a misleading conformational state of the channel. The presence of additional residues at the N-termini in the second structure probably blocks this interaction. It is therefore possible that the first structure has been perturbed by the detergent environment and crystal contacts (Jasti et al. Reference Jasti, Furukawa, Gonzales and Gouaux2007). In the light of a complementary structure of P2X receptor (discussed below), the second structure of ASCI (Gonzales et al. Reference Gonzales, Kawate and Gouaux2009) probably represents the functional desensitised state of the channel.

Fig. 5. Trimeric ion channels: (a) ASIC (3HGC), (b) P2X receptor (3H9V). Trimeric ion channels have thus far been found only in eukaryotes. Acid sensing ion channels (ASICs) and P2X receptors belong to the family of voltage-independent, ligand-gated cation channels. In contrast to the pentameric ligand-gated ion channels, ASIC and P2X receptor are trimeric. They have the commonly found architecture of two TM helices (blue, yellow and green) connected to a large extracellular domain (light green) that binds ligand. One salient feature in the extracellular domain of both these channels is the presence of a large number of cysteines that form disulphide bonds (red sticks), which is postulated to provide rigidity during conformational change upon ligand binding. The ligand in the case of ASCI is a proton, and for P2X it is ATP. The structures of ASIC and P2X receptor represent the closed states of the trimeric ion channels, since they have been crystallised at low pH and in the absence of ATP, respectively.
P2X4 Receptor
The concept of ATP as a signalling molecule was initially controversial. However, studies in sensory neurons revealed purinergic signalling to be a more common mechanism than previously thought. ATP synthesised in mitochondria is exported to the cytoplasm and then released to the extracellular space by membrane proteins that could either be channels or transporters. Extracellular ATP can either be hydrolyzed by extracellular enzymes or bind to one of many receptors that include the ionotropic P2X receptors (Schwiebert & Zsembery, Reference Schwiebert and Zsembery2003). Seven different subtypes have been found which can mix and match to form homo- and heteromeric receptors. Like many other membrane proteins P2X receptors, when overexpressed and purified, have a tendency to aggregate. Structure determination involved screening many different orthologues and successfully produced the structure of zebrafish P2X4 receptor (Kawate et al. Reference Kawate, Michel, Birdsong and Gouaux2009).
The extracellular domain of P2X4 receptor has a central rigid ‘body’ domain with a transthyretin-like β-sandwich motif around which other small domains are located (Fig. 5b). The upper region of this central domain makes contact with adjacent subunits while the lower region is devoid of any contact and is linked to the TM domain. The head domain, with a fold similar to oligosaccharide binding protein, is made up of three antiparallel β-strands and an α-helix that is located above the body domain. Other domains include the dorsal fin, and the right and left flipper. Numerous interactions are found between the body–body domains, head–body domains and the left flipper and dorsal fin of neighbouring subunits. It is speculated that such interactions between subdomains of the extracellular domain may play a role in the physiological properties and assembly of the P2X receptor subtypes. The present structure has been determined in the absence of agonist but, based on mutagenesis and the previously known ATP-binding site, an ATP binding motif has been proposed. Common features of P2X4 and ASCI extracellular domains include the presence of fenestrations at the membrane interface through which ions can pass, vestibules with acidic residues to hold cations and five disulphides that are also found in the extracellular domain of P2X4.
The TM domain of P2X4 has a similar architecture to the desensitised structure of functional ASCI (Gonzales et al. Reference Gonzales, Kawate and Gouaux2009; Kawate et al. Reference Kawate, Michel, Birdsong and Gouaux2009). In particular, the TM helices of P2X4 are antiparallel with an angle of 45° to membrane normal. The TM2 helices of the three monomers cross each other in the middle of the putative bilayer constricting the pore and are encircled by TM1. Comparison of the structures of P2X4 and ASCI also reveals a common principle on gating. The crossing of TM2 helices in both channels and the residues surrounding the constriction show some conservation but there must still be differences in the pores, since P2X4 channels are non-selective while ASCI channels are sodium selective.
Viral ion channels
Short peptides such as alamethicin and gramicidins that are synthesised non-ribosomally by multi-domain peptidyl synthases can form simple channels with antibacterial properties (Koglin & Walsh, Reference Koglin and Walsh2009). Some enveloped viruses also encode very small TM proteins that form channels and play a crucial role in their lifecycle. Proteins such as the M2 channel of influenza A virus, the Vpu channel of human immunodeficiency virus and the Kcv channel of algal chlorella virus PBCV-1 are ~100 amino acids in length and have been shown to function as channels (Fischer & Sansom, Reference Fischer and Sansom2002). The M2 channel of influenza A is the best characterised of these and forms a pH-gated proton channel. The infective cycle of influenza starts with the fusion of viral and host cell membranes. Endocytosis of the intact virus is followed by acidification within endosomes by cellular ATPases. The subsequent fusion of viral and endosomal membrane is mediated by an acid-induced conformational change in haemagglutinin that requires a change in the internal pH of the virion. The M2 channel is responsible for this change in pH, which allows the release of viral RNA into host cell. Drugs based on amantadine inhibit the pH change triggered by the M2 channel by inhibiting channel opening, but drug resistant mutants are on the increase.
Structures of the M2 channel have been determined by X-ray crystallography and NMR spectroscopy (Schnell & Chou, Reference Schnell and Chou2008; Stouffer et al. Reference Stouffer, Acharya, Salom, Levine, Di Costanzo, Soto, Tereshko, Nanda, Stayrook and Degrado2008). Neither of these structures is of a full-length protein. A short peptide corresponding to the TM region has been used for crystallography and a slightly longer version was used for NMR. Both structures reveal a tetrameric left-handed helical bundle. A narrow pore lined by hydroxyl and carbonyl groups from conserved residues is located at the N-terminus of the channel and is suggested to act as a solvent-filled path for proton transfer. Just below this pore region, the M2 channel is constricted by valine and then opens into an aqueous cavity lined by small residues. In the middle of the pore, a conserved histidine that acts as a pH sensor is in close contact with a tryptophan that is thought to act as a gate. Lower pH results in electrostatic repulsion of histidines and opening of the gate. The major difference between the X-ray and NMR structures is the mode of drug binding. In the X-ray structure a single drug molecule, amantidine, binds in the pore and blocks the channel. However, in the NMR structure four rimantadine drug molecules are found in the C-terminal region of the pore facing the bilayer, giving rise to speculation that the drugs may act by inhibiting the opening probability of the channel rather than directly blocking of the pore. Although both structures appear to be consistent with the mechanism of inhibition, with the drug-resistant mutations observed in the virus and with other biochemical observations, it is unclear if they reflect the true nature of the M2 channel since neither structure represents the full-length protein nor is their environment native. Hence there is always the possibility that detergent has perturbed their structures. Nevertheless, these structures represent a minimal ion channel.
Mechanosensitive channels
The ability of cells to sense a mechanical stimulus is achieved through specialised membrane proteins called mechanosensitive (MS) channels. In prokaryotes these mechanosensitive channels are used to respond to forces created by osmotic changes in the environment. In hyperosmotic conditions, accumulation of various solutes by dedicated transport systems offsets the efflux of water. In low osmotic conditions however, the influx of water generates a large turgor pressure that could potentially rupture the cell. MS channels in these cells act as safety valves and provide a quick defence against osmotic down shock by directly sensing the pressure from the lipid bilayer (Kung, Reference Kung2005). Homologues of prokaryotic MS channels are found in other cell-walled organisms such as fungi and plants. Much of our understanding of MS channels comes from studies in Escherichia coli. This bacterium possesses four MS channels: MscL (large conductance), MscS (small conductance), MscM (mini conductance) and MscK (regulated by potassium concentration) (Perozo & Rees, Reference Perozo and Rees2003). The presence of different MS channels allows the bacterium to respond to osmotic challenges of different magnitude. For example, MscL is activated close to the lytic limit of the lipid bilayer, while MscS is activated at slightly lower tension. MscL and MscS have been well-studied biochemically as well as structurally while much less is known about MscM and MscK.
Mechanosensitive channel, large
The crystal structure of MscL from Mycobacterium tuberculosis in a closed state reveals an oligomer of five subunits each with two TM helices and a C-terminal cytoplasmic domain (Chang et al. Reference Chang, Spencer, Lee, Barclay and Rees1998; Steinbacher et al. Reference Steinbacher, Bass, Strop and Rees2007). The pore forming TM1 helix is a tightly packed, right-handed α-helical bundle (Fig. 6a). It narrows towards the cytoplasm forming a hydrophobic constriction that could perform the role of a gate. TM2 wraps around the central TM1 and probably interacts with lipids on the outside. Each TM1 helix has four neighbours, consisting of two adjacent TM1s plus TM2 helices of its own and a neighbouring subunit. The periplasmic side of the pore helix TM1 is lined with polar residues. Although these are not conserved among the homologues, polar residues are always positioned along the permeation pathway. No interaction is observed between TM2 helices of adjacent subunits, which are presumably separated by lipid, and this loose packing might facilitate the necessary conformational change required during channel opening. A conserved amino acid stretch at the N-terminus adopts a helical conformation that is located near the surface of the membrane, inserting into a gap between TM1 and TM2 of neighbouring subunits. This short helix has been shown to play a role in MscL gating (Anishkin et al. Reference Anishkin, Chiang and Sukharev2005). The cytoplasmic C-terminal domain forms a left-handed helical bundle, which may act as a solute size pre-filter.

Fig. 6. Molecular architecture of prokaryotic mechanosensitive channels, side and top view: (a) MscL, closed (2OAR), (b) MscS, closed (2OAU), (c) MscS, open (2VV5). MscL and MscS are non-selective channels, activated in response to hypo-osmotic shock. MscL and MscS show unusually large conductances of 3 and 1 nS, respectively, which are much larger conductances than found in ion-selective channels because they make very large transient holes in the membrane. Although they carry out similar functions, the structures of these proteins are remarkably different indicating separate evolutionary pathways. MscL is a pentamer with two TM helices in each subunit, while MscS is a heptamer with three TM helices per monomer. One subunit in each channel is coloured in rainbow, blue at the N-terminus and red at the C-terminus. Despite their differences in oligomeric state, both proteins show a ring of single TM helices tightly packed to form the permeation pathway and covered by loosely packed outer helices. There is little sequence conservation between the pore forming helices of these two families of proteins. However, there is a striking common feature in the central helices of the two channels. When TM1 of MscL and TM3 of MscS are compared, there is a conserved pattern of alanine and glycine residues that allow tight packing of the pore forming helices, with interspersed hydrophobic residues that form the constriction pathway when the channels are closed. The presence of small amino acids in these helices must play a pivotal role in facilitating structural changes during gating. Indeed, the open structure of MscS shows a large rotation and tilting of helices, which results in the increase of pore diameter from 4·8 Å in the closed state to ~13 Å in the open state. Cross-linking and site-directed spin labelling studies indicate that MscL probably undergoes a similar conformational change to open the permeation pathway, creating a pore diameter of ~25 Å.
The recent structure of the MscL channel from Staphylococcus aureus reveals a tetramer, with two TM helices in each monomer (Liu et al. Reference Liu, Gandhi and Rees2009). It was essential to delete last 26 residues at the C-terminus to obtain crystals. The polypeptide conformation is slightly different from that in the MscL pentamer from M. tuberculosis. This structure of S. aureus MscL channel is thought to be in an intermediate expanded state when compared to the closed channel of M. tuberculosis. It is not obvious why these channels have different oligomeric structures.
Mechanosensitive channel, small
The crystal structure of from E. coli shows a heptamer with three TM helices and a large cytoplasmic domain in each subunit (Bass et al. Reference Bass, Strop, Barclay and Rees2002; Steinbacher et al. Reference Steinbacher, Bass, Strop and Rees2007). As opposed to MscL where both ends of the polypeptide are cytoplasmic, the N-terminus of MscS starts in the periplasm, followed by TM1 and TM2 that form outer helices enclosing the pore forming TM3 helices whose C-termini end in the cytoplasm (Fig. 6b). A glycine residue in TM3 produces a pronounced kink resulting in the C-terminus of TM3 being oriented nearly parallel to the membrane. TM3 interacts with symmetry related partners to form the permeation pathway. The N-terminus of TM3 is largely hydrophobic; of significant importance are the side chains of two leucine residues that block the pore. Thus, the permeation pathway in the observed structure is devoid of water and cannot pass ions; hence the structure of MscS probably reflects the closed state (Anishkin & Sukharev, Reference Anishkin and Sukharev2004). TM1 and TM2 within a subunit are antiparallel and show weak electron density possibly indicating their mobile nature. Two arginines found in TM1 and TM2 could explain the voltage modulation observed in single-channel recordings of MscS. The cytoplasmic region of MscS is fairly large and consists of two domains: a middle β-domain, where five short β-strands from each subunit pack together to form a continuous (35-stranded) β-sheet extending around the entire protein and a C-terminal domain that has mixed α/β structure. The C-terminal domain consists of a β-barrel formed by a single β-strand from each subunit, which is then surrounded by two α-helices that are packed against a three-stranded antiparallel β-sheet. The pore of MscS in the bilayer is accessible through seven openings each of 14 Å diameter formed by the cytoplasmic domains. An eighth opening passes through the centre of the C-terminal β-barrel, with a diameter of 8 Å. Such an intricate structure for the cytoplasmic domain may act as a pre-filtering device to limit the size of solutes that exit the cell during osmotic downshock.
Gating of MscL and MscS
Gating of both MscL and MscS is a complex process. Both channels are thought to exist in closed, open and inactive states. They also exhibit numerous subconductance states indicating multiple conformations. The response of MscS to changes in the voltage across the membrane in addition to membrane tension and its tendency to inactivate when the membrane tension increases slowly adds additional complexity (Akitake et al. Reference Akitake, Anishkin and Sukharev2005). In a biological context, the different gating behaviour of MscL and MscS has probably evolved for different conditions of stress. MscS opens when there is a small downshift in osmolarity and then inactivates once the turgor pressure is no longer threatening. MscL opens close to the lytic limit of membrane, so probably acts as a last resort when all other systems have failed.
Gating of MscL and MscS has been studied by techniques such as patch clamp and site-directed spin labelling with electron paramagnetic resonance (EPR) (Perozo et al. Reference Perozo, Cortes, Sompornpisut, Kloda and Martinac2002a; Akitake et al. Reference Akitake, Anishkin and Sukharev2005, Reference Akitake, Anishkin, Liu and Sukharev2007; Vasquez et al. Reference Vasquez, Sotomayor, Cordero-Morales, Schulten and Perozo2008). Different properties of lipids such as the chain length, intrinsic curvature of the membrane leaflet and fluidity of the membrane have been analysed with regard to their effects on gating of MS channels. Chain length and fluidity affect the threshold tension required to activate the channels, but do not trigger a conformational change. Addition of amphipathic molecules such as lysophosphatidyl choline (LPC) spontaneously opens the MS channels even in the absence of applied pressure indicating that lateral pressure mediated by lipids has a direct effect on the probability of channel opening (Perozo et al. Reference Perozo, Kloda, Cortes and Martinac2002b).
Recently, the crystal structure of an open conformation of MscS was obtained by mutating a single residue (from alanine to valine, A106V) near the N-terminus of TM3 helix (Wang et al. Reference Wang, Black, Edwards, Miller, Morrison, Bartlett, Dong, Naismith and Booth2008). In the closed MscS structure, this alanine packs against a glycine, G108, from a neighbouring subunit, but the presence of the bulkier valine must interfere with this interaction. Large changes are seen when the open structure is compared to the closed structure. The outer helices TM1 and TM2 are rotated by ~45° clockwise and are accompanied by a 15° tilt with respect to the seven fold axis in the open state compared with the closed state. In addition, the N-terminus of the pore forming helix TM3 is rotated by 15° clockwise while the C-terminus remains largely unchanged (Fig. 6c). Stabilisation of the open state is thought to occur through an interaction between the TM1-TM2 loops and both the C-terminus of TM3 of the same subunit and the N-terminus of TM3 of the neighbouring subunit. This change in tilt and rotation of the helices results in an ~8-Å increase of pore diameter upon channel opening to produce a pore with a diameter of ~13 Å. Such an increase in pore diameter by movement of helices has also been observed by site-directed spin labelling in liposomes, indicating that the open structure of MscS is not an artefact of detergent and crystallisation (Vasquez et al. Reference Vasquez, Sotomayor, Cordero-Morales, Schulten and Perozo2008). However, although the indirect deductions about solvent accessibility derived from modelling based on the site-directed spin labelling results agree well with the X-ray crystal structure observations for TM3, they do not agree for the other helices. Fig. 6 is therefore not the last word on the topic. Recent computational studies predict that the structure of the A106V mutant is a partially open state (Anishkin & Sukharev, Reference Anishkin and Sukharev2009).
There is no structure yet for an open conformation of a pentameric MscL but experiments by site-directed spin labelling, and by mutational and cross-linking analysis has allowed construction of a working model for its open state. As in MscS, tilting of both TM1 and TM2 helices are proposed, resulting in an increase of pore diameter to ~25 Å with only the pore forming TM1 helix, and not the outer TM2 helix, becoming exposed to the aqueous channel (Perozo et al. Reference Perozo, Cortes, Sompornpisut, Kloda and Martinac2002a). Despite the differences in the structure and gating behaviour, opening of the permeation pathway of both MscL and MscS can be explained by helix tilting. This is analogous to opening the iris in a camera, a mechanism that was proposed earlier based on cross-linking analysis and computational modelling (Sukharev et al. Reference Sukharev, Betanzos, Chiang and Guy2001). The essence of MS channel function lies in their ability to sense the pressure through the lipid bilayer. Hence it is surprising to learn that there is no structure yet of either MscL or MscS in lipid bilayers. It would be of great interest to see how the outer helices pack against lipids. A 3D crystal with a lipid bilayer would also provide an opportunity to study the effect of lateral pressure in situ and subsequent channel opening.
Eukaryotic MS channels
MS channels in animals reside in specialized organs such as the ear (hearing) and skin (touch) that detect mechanical stimuli and convert them into electrical signals, a process that has been termed ‘mechanotransduction’ (Gillespie & Walker, Reference Gillespie and Walker2001; Kung, Reference Kung2005; Sukharev & Corey, Reference Sukharev and Corey2004). Many biological process such as the twitching of nematode worms when touched, the response of plant roots and shoots to gravity, the determination of systemic osmolarity by circumventricular organs, or the sensing of blood pressure by baroreceptors in animals are all a result of mechanotransduction. MS channels respond to mechanical stimulus such as deformation of skin or oscillation of hair cells in hair bundles by rapid opening of the channels, resulting in a flow of ions that amplifies the signal. MS channels in eukaryotes interact with other proteins to form a mechanotransduction apparatus such as the ‘mec’ system of C. elegans (Chalfie, Reference Chalfie1997; Tavernarakis & Driscoll, Reference Tavernarakis and Driscoll1997). They are tethered to the extracellular matrix on one side and to the cytoskeleton in the cytoplasm, which allows the direct transfer of the signal and removes the need for secondary messengers. Some of the major membrane protein families in higher eukaryotes including the degenerin (DEG/ENaC) family, transient receptor potential (TRP) family and the two-pore domain K+-channel family have been proposed to be involved in mechanosensation (Gillespie & Walker, Reference Gillespie and Walker2001; Sukharev & Corey, Reference Sukharev and Corey2004; Kung, Reference Kung2005). The ASIC ion channels described in section 2.4 belong to the degenerin family, but there is no clear experimental evidence yet on whether they respond to mechanical stimuli. However, sequence homology and topological prediction of TM domains indicates that eukaryotic MS channels, for which there are no structures yet, may have a similar architecture to ASIC and share some gating properties with prokaryotic MS channels.
Aquaporins
Most biological membranes allow water transport by simple diffusion. However, the presence of specific water channels is crucial for many biological processes such as renal reabsorption, generation of aqueous humour in the eyes, secretion of sweat, saliva and tears, and regulation of cellular osmolarity in microbes and plants (Engel & Stahlberg, Reference Engel and Stahlberg2002; King et al. Reference King, Kozono and Agre2004). Hence, it is not surprising that one or more water channels, called ‘aquaporins’, are found in most organisms, principally to maintain water homeostasis. The 2003 Nobel Prize in Chemistry was awarded to Peter Agre for his role in discovering these channels (Agre, Reference Agre2004). Similar small molecules such as glycerol and urea are also transported through specific channels. Channels that transport glycerol as well as water are called aquaglyceroporins (Gonen & Walz, Reference Gonen and Walz2006) and show structural homology.
Since most ions are hydrated, an important function of water channels is to exclude passage of ions, especially protons, which would quickly short-circuit the electrochemical membrane potential. A continuous chain of hydrogen bonded water molecules can be an excellent proton pathway (de Grotthuss, Reference De Grotthuss1806). If aquaporins allowed proton permeation, there would be cellular acidification and collapse of the membrane potential. Thus, a water channel should have a mechanism to exclude protons while retaining the selectivity for a high flux (3×109 molecules/s) of water or similar molecules. This has been achieved in the aquaporin family by the juxtaposition of two short highly conserved sequences, both containing an asparagine–proline–alanine (NPA) motif, which together form the heart of the pore.
To date, 10 different aquaporin structures have been determined to high-resolution by X-ray or electron crystallography (Sui et al. Reference Sui, Han, Lee, Walian and Jap2001; Fu et al. Reference Fu, Libson, Miercke, Weitzman, Nollert, Krucinski and Stroud2000; Murata et al. Reference Murata, Mitsuoka, Hirai, Walz, Agre, Heymann, Engel and Fujiyoshi2000; Savage et al. Reference Savage, Egea, Robles-Colmenares, O'Connell and Stroud2003; Gonen et al. Reference Gonen, Sliz, Kistler, Cheng and Walz2004b; Harries et al. Reference Harries, Akhavan, Miercke, Khademi and Storud2004; Lee et al. Reference Lee, Kozono, Remis, Kitagawa, Agre and Stroud2005; Hiroaki et al. Reference Hiroaki, Tani, Kamegawa, Gyobu, Nishikawa, Suzuki, Walz, Sasaki, Mitsuoka, Kimura, Mizoguchi and Fujiyoshi2006; Tornroth-Horsefield et al. Reference Tornroth-Horsefield, Wang, Hedfalk, Johanson, Karlsson, Tajkhorshid, Neutze and Kjellbom2006; Horsefield et al. Reference Horsefield, Norden, Fellert, Backmark, Tornroth-Horsefield, Terwisscha van Scheltinga, Kvassman, Kjellbom, Johanson and Neutze2008; Newby et al. Reference Newby, O'Connell, Robles-Colmenares, Khademi, Miercke and Stroud2008; Fischer et al. Reference Fischer, Kosinska-Eriksson, Aponte-Santamaria, Palmgren, Geijer, Hedfalk, Hohmann, De Groot, Neutze and Lindkvist-Petersson2009). Aquaporins exist as tetramers in vitro as well as in vivo (Fig. 7a). The key architectural feature of the molecule is an internal duplication in which each half of the monomer consists of three TM helices and a re-entrant loop. The two repeats are related by an approximate twofold axis in the membrane plane giving the overall structure an unusual symmetry of inversion across the membrane. Each monomer is thus made up of a right-handed bundle of six TM helices (1–6) connected by five loops (A–E), which together form the channel. The two NPA motifs are located at the N-termini of the re-entrant loops B and E, which fold into the protein from opposite sides to meet in the middle of the bilayer. The C-terminal halves of these loops form short helices (HB and HE), which are roughly aligned and together can be considered to form a kinked seventh TM helix (Fig. 7b) with a central discontinuity. Another conserved feature in the aquaporin family is the aromatic/arginine (ar/R) constriction site, in which arginine and one or more aromatic residues usually play a crucial role in selectivity and ion exclusion. Varying numbers of water molecules (3–9) have been observed in the pore in different structures, depending on the open or closed state of the channel. Pore diameter at the entrance of a pure water channel is 3 Å, slightly larger than a water molecule, while that of a glycerol channel is 3·8 Å.

Fig. 7. (a) Tetramer of aquaporin (2B60). (top) Aqp0, determined by electron crystallography viewed from the extracellular side, shows an individual water channel in each monomer with water molecules depicted as red spheres. Lipids probably fill the cavity at the centre of the tetramer. So far only in the structure of human Aqp5, has an ordered lipid molecule been identified in this region (Horsefield et al. Reference Horsefield, Norden, Fellert, Backmark, Tornroth-Horsefield, Terwisscha van Scheltinga, Kvassman, Kjellbom, Johanson and Neutze2008). (b) Monomer of aquaporin showing how it has evolved by gene duplication. The N and C terminal halves (blue and green) of each polypeptide are related by a pseudo twofold symmetry axis, which is parallel to the membrane plane and perpendicular to the page. Thus, the two halves of the molecules have opposite orientations in the membrane. Loop C (magenta) on the extracellular side connects the two halves. The conserved NPA motifs and short helices (HB and HE) are depicted in yellow. Three water molecules (red spheres) as seen in the EM structure of Aqp0 are shown along the pore region (Gonen et al. Reference Gonen, Sliz, Kistler, Cheng and Walz2004b). The hourglass like structure of aquaporin was predicted based on primary sequence and biochemical analysis years before the first structure was determined (Jung et al. Reference Jung, Preston, Smith, Guggino and Agre1994).
Substrate selectivity can be understood by comparing different aquaporin structures. Substitution of residues around the ar/R constriction site forms the basis of permeability and selectivity. Pure water conducting channels such as Aqp1 and AqpZ have two charged residues (arginine and histidine) along with a polar residue and a phenylalanine (Sui et al. Reference Sui, Han, Lee, Walian and Jap2001; Savage et al. Reference Savage, Egea, Robles-Colmenares, O'Connell and Stroud2003). In the homologous glycerol facilitator GlpF, histidine is replaced by glycine, phenylalanine by tyrosine and the polar residue by hydrophobic phenylalanine providing an environment that accommodates glycerol. Thus, GlpF is highly permeable to glycerol but less so to water (Fu et al. Reference Fu, Libson, Miercke, Weitzman, Nollert, Krucinski and Stroud2000). Channels such as the archeal AqpM that conduct glycerol poorly in vitro tend to have an aliphatic substitution for histidine (Lee et al. Reference Lee, Kozono, Remis, Kitagawa, Agre and Stroud2005).
Based on the structure and subsequent molecular dynamics (MD) simulations, proton exclusion is explained by a ‘hydrogen bond isolation mechanism’ (Murata et al. Reference Murata, Mitsuoka, Hirai, Walz, Agre, Heymann, Engel and Fujiyoshi2000; de Groot & Grubmuller, Reference De Groot and Grubmuller2001; Tajkhorshid et al. Reference Tajkhorshid, Nollert, Jensen, Miercke, O'Connell, Stroud and Schulten2002). The side chains of the asparagines in the NPA motif extend into the pore and the positive end of the dipole moments of the half helices are focused on these amido groups. The oxygen lone pairs of the central water molecule accept hydrogen bonds from the amide hydrogen atoms of the two asparagine side chains. This orients the two hydrogens in the water molecule perpendicular to the pore axis, thereby preventing the formation of hydrogen bonds with neighbouring water molecules and dividing the pore into halves, ensuring that only water and not hydronium ions can bind to this central site. MD simulations show that the lines of water molecules in the two halves of the pore have opposite hydrogen bond polarity thereby preventing the passage of protons. It is interesting to note that mutation of arginine in the ar/R constriction site is sufficient to allow some protons to leak through (Beitz et al. Reference Beitz, Wu, Holm, Schultz and Zeuthen2006). Thus, the positive charge in the constriction site and the positive dipole at the centre of the pore together form an electrostatic barrier for protons.
Regulation of some aquaporins involves the loop regions. It is achieved at various levels by pH, phosphorylation, calcium or hormones. Human aquaporins such as Aqp0, Aqp3 and Aqp6 have been shown to be pH sensitive. The lens-specific, junction-forming Aqp0 deserves special mention because it undergoes channel closure by cleavage at the N-terminus, followed by a conformational change in residues lining the pore (Gonen et al. Reference Gonen, Cheng, Kistler and Walz2004a). A similar scenario is observed in yeast aquaporin where the N-terminus of the molecule acts as a gate (Fischer et al. Reference Fischer, Kosinska-Eriksson, Aponte-Santamaria, Palmgren, Geijer, Hedfalk, Hohmann, De Groot, Neutze and Lindkvist-Petersson2009). Phosphorylation of conserved serine residues and protonation of histidine in plant aquaporin SoPIP2 regulates the water conductance. The structure of SoPIP2 solved in open and closed conformation revealed a role for loop D and helix 5 in water conductance (Tornroth-Horsefield et al. Reference Tornroth-Horsefield, Wang, Hedfalk, Johanson, Karlsson, Tajkhorshid, Neutze and Kjellbom2006).
Aquaporins are one of the families of membrane protein whose structure has been determined at high resolution both in detergent micelles and in lipid bilayers (Gonen et al. Reference Gonen, Sliz, Kistler, Cheng and Walz2004b; Harries et al. Reference Harries, Akhavan, Miercke, Khademi and Storud2004). Although all aquaporins adopt the same fold, subtle differences observed in these structures have increased our understanding of how the channels work. Questions remain about how some aquaporins allow anion permeation and about the mechanisms behind regulation. Finally, there is a very recent new structure of a formate transporter whose fold closely resembles aquaporin, yet form a symmetric pentamer rather than a symmetric tetramer (Wang et al. Reference Wang, Huang, Wang, Cheng, Huang, Lu, Xu, Wang, Yan and Shi2009)
Ammonia channel
Assimilation of nitrogen is essential for all biological processes. Nitrogen can be taken up in different forms ranging from ammonia, nitrate and nitrite to atmospheric nitrogen. Most organisms prefer ammonia since it can be directly assimilated into glutamine with the least energetic cost while inorganic sources such as nitrate or nitrite need to be reduced before they can be incorporated into the metabolic pathway. In aqueous solution, ammonia (NH3) is a weak base that exists in equilibrium with ammonium ions with a pK a of 9·25. Most biological membranes are sufficiently permeable to ammonia for growth of microbes but in an environment with low concentrations of ammonia, dedicated transport system have evolved to meet the needs of the cell (Khademi & Stroud, Reference Khademi and Stroud2006). In prokaryotes and plants, these are called ammonia transporters (Amt). In yeast, they are methylammonium permeases (MEPs) and, in animals, they include the Rh family of proteins. Expression of Rh proteins is observed in many tissues such as liver, kidney and skin and they play a major role in acid homeostasis by removing ammonia from the blood (Ludewig, Reference Ludewig2006). The whole family of ammonia channels is now known as the Amt/MEP/Rh family.
Crystal structures of the ammonium channel from E. coli (Khademi et al. Reference Khademi, O'Connell, Remis, Robles-Colmenares, Miercke and Stroud2004; Zheng et al. Reference Zheng, Kostrewa, Berneche, Winkler and Li2004), Archaeoglobus fulgidus (Andrade et al. Reference Andrade, Dickmanns, Ficner and Einsle2005) and Nitrosomonas europaea (Lupo et al. Reference Lupo, Li, Durand, Tomizaki, Cherif-Zahar, Matassi, Merrick and Winkler2007) have been determined by X-ray crystallography (Fig. 8a). Ammonium transporters form a trimer with a separate pathway in each monomer for substrate transport. Each monomer comprises 11 TM helices arranged in a right-handed α-helical bundle. Helices 1–5 and 6–10 are related to each other by a pseudo twofold axis that lies in the membrane plane (Fig. 8b), giving rise to the same type of inverted symmetry as in the aquaporins. A long, straight helix (TM11) lies at 45° to the membrane plane covering each monomer. TM1 from each monomer packs tightly at the periplasmic/extracellular end of the threefold axis to seal the passage while tapering towards the cytoplasmic end to leave an open pocket. The channel in each monomer is hydrophobic with polar cytoplasmic and extracellular vestibules. The vestibules at the extracellular side expose numerous carbonyl groups that help to attract ammonium ions and water.

Fig. 8. Ammonia channel (1U7G). (a) Trimeric ammonia channel, viewed from the extracellular side, showing an ammonium ion (red) at the periplasmic entrance and three ammonia molecules (yellow) in the lumen. One monomer coloured as in (b) clearly shows the pseudo twofold symmetry. The trimer of the ammonia channel is very stable even in the presence of SDS and tight packing between monomers is possibly the reason why the crystals are so well ordered. Among the membrane protein structures known so far, this eleven helical bundle is unique to the ammonia channel family. (b) Monomer of the ammonia channel: Unlike the aquaporins, the sequence of AmtB does not reveal any hint of gene duplication, but the structure shows that helices 1–5 (blue) and 6–10 (green) are related by pseudo twofold symmetry. Like aquaporins the pseudo twofold axis lies in the membrane plane so that the two halves of the molecule have opposite orientations in the membrane. Together they make the pore. Helix 11 (magenta) lies at a 45° angle to the membrane and makes contact with both halves of the monomer.
Ammonia channels are functionally electroneutral. It was thought initially that ammonium ions and protons were counter-transported. However, the structures show rather passive channels through which neutral ammonia is likely to pass. Structures solved in the presence of ammonia or methylammonia reveal weak electron densities at four different sites (Khademi et al. Reference Khademi, O'Connell, Remis, Robles-Colmenares, Miercke and Stroud2004). Only the first site at the periplasmic entrance of the pore contains an ammonium ion while the other three found in the lumen are occupied by ammonia. The entrance to the hydrophobic channel is very narrow and constricted by two phenylalanines, a tryptophan and serine. An incoming ammonium ion interacts with aromatic rings via cation-π interactions and hydrogen bonds with a serine side chain. This interaction results in rotation of the indole ring of tryptophan, which exposes a buried aspartate. This conserved aspartate is thought to act as a proton acceptor via water molecules and mutation of this residue renders the channel inactive. Two phenylalanines act as gates to let ammonia through, but block extracellular solvent from the pore (Lin et al. Reference Lin, Cao and Mo2006).
The lumen of the pore is ~20 Å in length, devoid of any water molecules and lined predominantly by aliphatic non-polar side chains. The hydrophobic nature of the pore has been confirmed by solving the structure of AmtB in the presence of xenon, which shows two Xe binding sites in the pore (Andrade et al. Reference Andrade, Dickmanns, Ficner and Einsle2005). Two conserved histidines (His 168 and 318 in E. coli AmtB) lie adjacent to the ammonia sites in the pore and are hydrogen bonded to one another in the mid-membrane. Mutation to any other amino acid (except for H168E in E. coli, which is partially active) abolishes conduction and so implies a specific role for these histidines in transport (Javelle et al. Reference Javelle, Lupo, Zheng, Li, Winkler and Merrick2006). In the crystal structures, ammonia molecules form weak hydrogen bonds to the histidines and such weak interactions may allow for rapid diffusion of the ammonia molecules through the pore. On leaving the pore, ammonia is reprotonated at the cytoplasmic side since its pK a is above 9. Water molecules are not transported and even in the absence of ammonia, the crystal structures do not show any water molecules in the lumen (Khademi et al. Reference Khademi, O'Connell, Remis, Robles-Colmenares, Miercke and Stroud2004). By transporting neutral ammonia, the channels also exclude leakage of similar sized K+ ions which if permitted would affect the membrane potential.
In prokaryotes and plants, proteins belonging to the PII signal transduction family regulate ammonia transport (Arcondeguy et al. Reference Arcondeguy, Jack and Merrick2001). Often, the ammonia transport gene is linked with its regulator GlnK, a soluble trimeric protein that binds to the ammonia channel on its cytoplasmic face and blocks transport (Conroy et al. Reference Conroy, Durand, Lupo, Li, Bullough, Winkler and Merrick2007; Gruswitz et al. Reference Gruswitz, O'Connell and Stroud2007). GlnK has a multi-functional role since it can also bind to glutamate synthase, the prime acceptor of ammonia. In nitrogen limiting conditions, the concentration of 2-oxoglutarate (2-KG) in the cell rises and binds to GlnK in the presence of ATP and magnesium. This leads to the dissociation of GlnK from Amt and opening of the channel. When intracellular nitrogen levels are relatively high, the level of 2-KG drops and GlnK can now bind back to Amt, blocking ammonia uptake. The trimeric architecture of Amt channel may have evolved to match the symmetry of the regulatory proteins.
Active transport
Active transporters use energy for transporting molecules and ions against a concentration gradient. They are classified as primary or secondary transporters (Mitchell, Reference Mitchell1967). When the source of energy is derived from ATP or light or redox energy, these transporters are called primary transporters, which are predominantly involved in maintaining ion gradients across the membrane. Secondary active transporters utilise the free energy stored in an electrochemical ion gradient to drive the translocation of substrates against their concentration gradient. Primary and secondary transport processes interact via the membrane potential. Thus, ion gradients generated from chemical, redox or light energy are used in many diverse processes such as drug extrusion, flagellar rotation or metabolite transport. Most cells maintain electrochemical gradients of both H+ and Na+ ions across the cytoplasmic membrane by primary or secondary ion translocation processes. In many organisms, the proton motive force generated by substrate oxidation via the respiratory chain is used to extrude Na+ from the cell by a Na+/H+ antiport mechanism, thereby establishing an electrochemical Na+ gradient across the cytoplasmic membrane called ‘smf’ (sodium motive force). Alternatively, a variety of prokaryotic cells possess primary Na+ pumps that can also generate smf.
Both primary and secondary transporters can be classified into uniporters, symporters or antiporters based on the characteristics of substrate transport. The mechanism of translocation in all these transporters can be explained by an alternating access mechanism where access to a ligand binding site in the protein alternates between two sides of the membrane for uptake and release of substrate (Jardetzky, Reference Jardetzky1966). Control of coupled reaction sequences in active transport depends on sequential changes in the protein structure as the reaction proceeds. These changes are brought about by specific interactions with the substrate, the binding forces being used to stabilise a conformation with altered properties or a transition state between successive conformations.
Primary transporters
P-type ATPase
Membrane proteins that are transiently phosphorylated as a key step in the transport cycle are called P-type ATPases. They utilise chemical energy from the hydrolysis of ATP to pump ions against a concentration gradient. In animal, plant and fungal cell membranes, P-type ATPases play a major role in the generation of a membrane potential, which is subsequently used by ion channels and secondary transporters. Other notable biological functions include the control of cytoplasmic calcium concentrations by the sarcoplasmic reticulum Ca2+-ATPase (SERCA) that is involved in skeletal muscle contraction and acidification of the stomach by the gastric H+/K+-ATPase.
P-type ATPases are distributed in all organisms and have been divided into five distinct classes (Type I–V) based on sequence homology. They transport a wide range of common cations such as H+, Na+, K+, Ca2+ and Mg2+ and also have roles in the efflux of heavy metals (Kuhlbrandt, Reference Kuhlbrandt2004). All P-type ATPases have their cytoplasmic domains subdivided into phosphorylation (P), nucleotide binding (N), and actuator (A) domains. The P domain forms the catalytic core with a Rossmann fold, which harbours a conserved aspartate that becomes phosphorylated during the transport cycle. The N domain is inserted within the P domain and is linked to it by a conserved hinge of two antiparallel strands. The N domain is the least conserved of all three cytoplasmic domains. It has variable size and it is responsible for the delivery of phosphate from ATP to the P domain. A conserved ‘TGE’ motif in the A domain plays a role both in the hydrolysis of phosphate from the P domain aspartate and in the gating mechanism.
The TM domain of P-type ATPases consists of as few as six TM helices (E. coli Kdp ATPase), or eight in the case of heavy metal pumps but they more generally contain 10 TM helices (M1 to M10). As one would expect, the TM domain is the least conserved region in the P-type ATPase family since it has evolved to coordinate and transport a variety of ions of different size and charge (Fig. 9). The A domain is inserted between TM helices M2 and M3 while the P domain is inserted between M4 and M5. In fact, helices M2 and M5 extend a long way from the membrane and interact with the cytoplasmic domains. TM helices M7 to M10 play an anchoring role with a specialised function in each subgroup. For example, in sodium pumps (Na+/K+-ATPase) they interact with β- and γ-subunits, although these additional subunits are absent in heavy metal pumps. The overall architecture of P-type ATPases is well conserved as revealed from the crystal structures of three different classes namely Na+/K+-ATPase, H+-ATPase and Ca2+-ATPase (Toyoshima et al. Reference Toyoshima, Nakasako, Nomura and Ogawa2000; Morth et al. Reference Morth, Pedersen, Toustruo-Jensen, Sorensen, Petersen, Andersen, Vilsen and Nissen2007; Pedersen et al. Reference Pedersen, Buch-Pedersen, Morth, Palmgren and Nissen2007; Shinoda et al. Reference Shinoda, Ogawa, Cornelius and Toyoshima2009). Even the TM domains that show very little sequence homology have remarkably similar structures with differences matched to their different ion transport specificity. In the H+-ATPase, only a single aspartate is found in the TM region, while the calcium and sodium pumps have multiple buried acidic residues that coordinate the larger ions they transport. Of particular interest are helices in all three ATPase structures that are partially unwound within the membrane-spanning region (M4 and M6 in Ca2+-ATPase), exposing main chain carbonyl and amides that coordinate cations, a recurring theme observed in many ion transport proteins. Such discontinuous helices create a cavity that is designed to coordinate ions and associated solvating water molecules. The structure of the Na+/K+-ATPase with its additional regulatory (β and γ) subunits shows how the helices M7 to M10 interact with other proteins (Fig. 9b).

Fig. 9. Molecular architecture of P-type ATPases shown with the cytoplasmic active site at the bottom, using the convention adopted throughout this review. (Note that in the P-type ATPase field, the orientation is normally inverted from that shown here). (a) Ca2+-ATPase (2ZBD) and (b) Na+/K+-ATPase (2ZXE). The formation of an acid-stable phosphorylated intermediate during the transport cycle of this class of membrane proteins gives rise to their name, P-type ATPase. All proteins in this family have three cytoplasmic domains, a nucleotide binding domain (green), a phosphorylation domain (yellow) and an actuator domain (blue). The TM domain (brown) with 10 α-helices is the least conserved of all domains. The β-subunit of Na+/K+-ATPase is shown in pink and the γ-subunit (FXYD protein) in orange. The transport cycle of P-type ATPases has been described by Post and Albers (Albers et al. Reference Albers, Fahn and Koval1963; Post et al. Reference Post, Sen and Rosenthal1965) to alternate between E1 and E2 states with different affinities for ions and nucleotides. Two representative structures of Ca2+-ATPase and Na+/K+-ATPase show a general architecture preserved in P-type ATPases. It has been possible to trap intermediates in the transport cycle by the use of transition state analogues and inhibitors with ATPase. The E1P state of Ca2+ ATPase (a) with calcium ions (cyan) occluded within the protein was obtained by the addition of AlF4− (aquamarine) and Mg.ADP (orange). In the Na+/K+-ATPase (b), when a phosphate mimic such as MgF42− (magenta) is crystallised with counterion potassium (purple), it was possible to trap the pump in the ion-occluded E2 state. For a complete discussion of multiple structures and conformational changes, see a recent review (Toyoshima, Reference Toyoshima2008).
Ion pumps have evolved to build ion gradients. Hence, it is crucial that pumps do not leak. It is thought that pumps must have two gates that open alternately as opposed to a single gate in ion channels. At any given time, only one of the ion pump gates is open. It is also possible that there is an intermediate state in which both gates are closed and the ions are occluded at one point in the transport cycle. Structural changes required to open and close the gates sequentially limit the rate of ion transport to ~100 ions/s as opposed to 107 ions/s in ion channels where the single gate is either open or closed (Gadsby et al. Reference Gadsby, Takeuchi, Artigas and Reyes2009). The transport cycle of P-type ATPases follows an alternating access mechanism that was described initially for the Na+/K+-ATPase. It is known as the Post-Albers scheme (Albers et al. Reference Albers, Fahn and Koval1963; Post et al. Reference Post, Sen and Rosenthal1965) but is applicable to all members of the P-type ATPase family. The pump alternates between two conformations (E1 and E2) with different affinities for ions and ATP. Binding of ion-1 (Na+ in the Na+/K+-ATPase) from the cytoplasm to a high affinity site in the E1-state stimulates phosphorylation of the pump by ATP, resulting in an E1-P state in which the ion is occluded from both sides. Release of ADP and a subsequent conformational change results in an E2-P state with reduced affinity for ion-1 that is then released to the extracellular surface or vesicle lumen. The hydrolysis of phosphate is stimulated when ion-2 binds to the site vacated by ion-1 (K+ in the Na+/K+-ATPase) resulting in an ion occluded E2 state. The pump reverts back to the E1 state releasing ion-2 into the cytoplasm. Apart from the plant and fungal plasma membrane H+-ATPases, which are electrogenic and transport only protons, all other P-type ATPases transport counter-ions. The number of ions transported per catalytic cycle differs in each pump. Generally an asymmetry is observed such as three Na+ for two K+ in the Na+/K+-ATPase or two Ca2+ for probably three H+ in the Ca2+-ATPase, so there is a degree of electrogenicity in all P-type ATPases.
Since the identification of the sodium pump by Jens Skou (Skou, Reference Skou1957), much of the structural research on P-type ATPases has been dedicated to understanding the different conformations of the pumps as described in the Post-Albers scheme. The structure of the Ca2+-ATPase determined by X-ray crystallography revealed for the first time the architecture of a P-type ATPase (Toyoshima et al. Reference Toyoshima, Nakasako, Nomura and Ogawa2000). Subsequently in the last few years as many as 22 structures of the Ca2+-ATPase trapped with transition state analogues or inhibitors have provided us with an almost complete picture of the transport cycle of a P-type ATPase (Toyoshima, Reference Toyoshima2008). Two calcium-binding sites in the TM region (sites I and II) have been identified from the structure of the Ca2+-ATPase. Site II is located closer to the cytoplasmic face while site I is deeper within the TM domain. Calcium binding shows co-operative behaviour. Ion binding at site I is essential for converting site II into a high affinity-binding site. The calcium binding in site I is coordinated by side chain carbonyl groups and water while site II is made up mainly of main chain carbonyl groups from the unwound TM helices with a single aspartate residue contributing to both sites.
Comparison of multiple structures in different states reveal that the cytoplasmic (P, N and A) domains move as rigid bodies during the transport cycle communicating the phosphorylation event in the P domain to the TM domain through the extended helices M2 and M5. Much of the conformational change in the TM domain is centred on M5, which forms the backbone. While the helices M7 to M10 remain unchanged, substantial movement is observed in M1 to M6. Partially unwound helices remain so throughout the transport cycle. The affinity of the calcium binding sites is modulated by changes occurring in TM domains coupled to movements of the cytoplasmic domain. This results in the binding of Ca2+ in the E1 state or its release in E2 state. Based on the structures, it has been possible to explain how events occurring at different sites (phosphorylation/dephosphorylation in the cytoplasmic domain and binding of calcium in the TM domain) separated by ~50 Å may be coupled mechanically.
Light-driven pumps
Membrane proteins of the bacteriorhodopsin family have a variety of functions, all triggered by light absorption and isomerisation of the chromophore retinal. Bacteriorhodopsin, the founding member of this family, was discovered by Stoeckenius & Rowen (Reference Stoeckenius and Rowen1967) in the purple membrane from Halobacteria, and subsequently shown to function as a light-driven proton pump (Oesterhelt & Stoeckenius, Reference Oesterhelt and Stoeckenius1973). It was shown to be a seven-helix bundle (Henderson & Unwin, Reference Henderson and Unwin1975) and since then many related structures have been solved at atomic resolution by both X-ray and electron crystallography (Belrhali et al. Reference Belrhali, Nollert, Royant, Menzel, Rosenbusch, Landau and Pebay-Peyroula1999; Luecke et al. Reference Luecke, Schobert, Richter, Cartailler and Lanyi1999)
Members of the family are widely distributed in organisms from bacteria and archaebacteria up to simple eukaryotes such as Neurospora (Bieszke et al. Reference Bieszke, Spudich, Scott, Borkovich and Spudich1999) and Chlamydomonas (Nagel et al. Reference Nagel, Ollig, Fuhrmann, Kateriya, Musti, Bamberg and Hegemann2002). Their functions include proton pumping, chloride ion pumping (Schobert & Lanyi, Reference Schobert and Lanyi1982) and phototactic signalling (Gordeliy et al. Reference Gordeliy, Labahn, Moukhametzianov, Efremov, Granzin, Schlesinger, Buldt, Savopol, Scheidig, Klare and Engelhard2002). All members of the family have a similar photocycle initiated by the isomerisation of all-trans retinal to 13-cis retinal. A sequence of spectroscopically distinct intermediates coupled to the particular function follows. In bacteriorhodopsin, the key intermediate is M412 from which a proton has been released on the extracellular side, but the Schiff base is not deprotonated in the halorhodopsin photocycle nor in the sensory rhodopsins. Bacteriorhodopsin (bR) and sensory rhodopsin (sRII) both show a substantial structural change that is important for ion pumping or signalling via interaction with a transducer protein. In bacteriorhodopsin, the largest structural change consists of 3- to 4-Å movements of the cytoplasmic end of helix 6 and a smaller movement of helix 7 during the second half of the photocycle (Fig. 10a), which opens up the cytoplasmic half-channel to allow reprotonation of the Schiff base from the cytoplasmic surface (Subramaniam & Henderson, Reference Subramaniam and Henderson2000). This structural change is likely to be common to all members of the family, though coupled in slightly different ways to the different functions. The most complex structure solved so far is the Natronobacter sRII:HtrII complex (Gordeliy et al. Reference Gordeliy, Labahn, Moukhametzianov, Efremov, Granzin, Schlesinger, Buldt, Savopol, Scheidig, Klare and Engelhard2002), which is a dimer with a twofold axis relating the two HtrII helical hairpins and the two molecules of sRII bound on each side. The light-induced structural change in sRII helix F is thought to cause a helix rotation in the HtrII dimer, which is then coupled to the methyl-accepting cytoplasmic domain and subsequently to the photophobic behavioural response of the bacteria (Fig. 10b). A recent addition to the family comes from the discovery of channelopsin (Nagel et al. Reference Nagel, Ollig, Fuhrmann, Kateriya, Musti, Bamberg and Hegemann2002), which is a light-activated ion channel that is proving to be a useful tool in electrophysiology, but whose structure has not yet been determined.

Fig. 10. Light driven pumps: (a) bR (1FBB and 1FBK) and (b) sRII:HtrII dimeric complex (1H2S). Bacteriorhodopsin is one of the simplest monomeric membrane proteins, composed of a seven-TM helix bundle with the chromophore retinal (red) bound in the centre of the bundle. Light isomerises the retinal and a proton is then released towards the extracellular side from its Schiff base with K216 (blue). A large structural change, (from purple to yellow in panel a) then opens the cytoplasmic half-channel to allow reprotonation of the Schiff base and completion of the cycle. A similar structural change is thought to be present in other members of the bacteriorhodopsin family. Here the structure of sRII, a photophobic sensor (orange) in halobacteria is shown in complex with the helical hairpin dimer (green) of its cognate transduction partner, HtrII. In this case, the structural change couples to a cytoplasmic chemotactic/phototactic methylation cascade.
ATP synthases
In the inner membranes of prokaryotes, mitochondria and chloroplasts, the energy stored in electrochemical gradients can be converted first into subunit conformational changes by mechanical rotation in the F1Fo-ATPases, and these conformational changes then catalyse the synthesis of ATP (Nakamoto et al. Reference Nakamoto, Baylis Scanlon and Al-Shawi2008). In eukaryotes, vacuolar V1Vo-ATPases generate a proton motive force (pmf) by hydrolysing ATP to cause acidification of the internal space of vacuoles (Drory & Nelson, Reference Drory and Nelson2006; Jefferies et al. Reference Jefferies, Cipriano and Forgac2008). The archeal A1Ao-ATPase is more closely related to eukaryotic V-ATPases but its normal function is to synthesise rather than hydrolyze ATP (Gruber & Marshansky, Reference Gruber and Marshansky2008). The basic architecture of all these ATPases includes a soluble catalytic domain (F1, V1 or A1) and a membrane embedded rotor domain (Fo, Vo or Ao) linked by central and peripheral stalks. The number of subunits associated with the catalytic domain is constant but the number of subunits associated with Fo, Vo or Ao and the peripheral stalk varies within each class of ATPase and depends on the organism from which it is isolated. Perhaps this complexity is one reason why a high-resolution structure of a complete ATPase of any class has not yet been reported, although a medium resolution X-ray structure of yeast F1c10-ATPase (Stock et al. Reference Stock, Leslie and Walker1999), which forms a major part of the whole structure, shows the organisation of the c-ring with respect to the soluble F1 domain. In addition, structures determined by single particle cryo-EM of intact bovine F1Fo-ATPase (Rubinstein et al. Reference Rubinstein, Walker and Henderson2003) and tobacco hornworm V1Vo-ATPase (Muench et al. Reference Muench, Huss, Song, Phillips, Wieczorek, Trinick and Harrison2009) have provided molecular envelopes into which high-resolution subunit structures have been docked (Dickson et al. Reference Dickson, Silvester, Fearnley, Leslie and Walker2006). Several reviews describing the molecular architecture and mechanism of F-type and V-type ATPases are available (Drory & Nelson, Reference Drory and Nelson2006; Gruber & Marshansky, Reference Gruber and Marshansky2008; Nakamoto et al. Reference Nakamoto, Baylis Scanlon and Al-Shawi2008). Our focus here will be on the membrane embedded rotor domain.
The membrane embedded rotor domain in F1Fo-ATPases consists of a minimal core of the three TM proteins a, b and c such as found in E. coli but additional subunits are sometimes found in ATPases from different organisms. Subunit a, predicted to span the membrane five times harbours a crucial arginine essential for ion transport. Subunit b, which contains a single TM helix in the bacterial enzyme (but two in mitochondria), forms the peripheral stalk that interacts with subunit δ of the F1 subunit (or OSCP, d and F6 in mitochondria). Subunit c, also called proteolipid protein or c-ring, forms a circular oligomer that interacts with subunit a of the rotor domain and the F1 domain through the central stalk. Of the membrane embedded proteins, only the structure of subunit c has been solved by X-ray crystallography. There is a sodium dependent F1Fo-ATPase from Ilyobacter tartaricus (Meier et al. Reference Meier, Polzer, Diedrichs, Welte and Dimroth2005), a proton dependent F1Fo-ATPase from chloroplast (Pogoryelov et al. Reference Pogoryelov, Yildiz, Faraldo-Gomez and Meier2009; Vollmar et al. Reference Vollmar, Schlieper, Winn, Buchner and Groth2009) and an NMR spectroscopic study on monomeric subunits of E. coli c-ring in organic solvent (Girvin et al. Reference Girvin, Rastogi, Abildgaard, Markley and Fillingame1998). Subunit c of I. tartaricus is an 11-membered ring while the chloroplast ring has 14 subunits. In both cases each subunit is made up of a helical hairpin with a conserved glutamate approximately in the middle of the bilayer (Fig. 11b).

Fig. 11. Side and top view of the rotor subunits from prokaryotic V-type or F-type ATPases: (a) K-ring (2BL2) and (b) c-ring (1YCE). The rotors of the F1Fo and V1Vo-ATPases comprise multimers of identical subunits that give rise to c-rings and K-rings, respectively. The c-ring of Ilyobacter tartaricus is an 11-membered ring in which each monomer is a helical hairpin (two subunits are highlighted in cyan) with a conserved glutamate or aspartate residue (red sticks) in the middle of membrane. The K-ring of Enterobacter hirae is a 10-membered ring, with each monomer having two homologous hairpins (cyan and yellow) covalently linked resulting in four TM helices per protein monomer. Only one of these hairpins has the conserved glutamate (red sticks in subunit coloured yellow) resulting in a lower ion to ATP ratio than would be obtained if both hairpins retained a glutamate. However, the structure is equivalent to a 20-membered c-ring. Both proteins are sodium-dependent ATPases and the crystal structure shows bound sodium ions (purple) in all subunits. Residues from the same subunit mediate sodium coordination in K-ring, whereas in the c-ring residues from neighbouring subunit contribute to sodium coordination. The top view of both proteins shows a central hole filled by lipids (green sticks).
The rotor domain of the Vo-ATPase has a minimal core of only two subunits, a and c. The structure of the c subunit of V-ATPase from Enterobacter hirae (Murata et al. Reference Murata, Yamato, Kakinuma, Leslie and Walker2005) has 10 subunits each with four TM helices (which form the K-ring) rather than two helices found in F-ATPases and shows evidence of gene duplication. The four TM helices form two covalently linked and homologous hairpins (Fig. 11a). However, during the process of duplication and fusion, only one hairpin has retained the conserved glutamate, a feature that results in a lower ion:ATP ratio than it would have if both hairpins retained the glutamate. The lower ratio also makes it likely that these enzymes have evolved to become proton pumps that can only hydrolyse ATP. Ion translocation in both F-type and V-type ATPase has been postulated to occur through two half channels formed by subunit c and subunit a, via the following hypothetical mechanism. Rotation of the c ring brings the conserved glutamate in subunit c close to the conserved arginine in subunit a, which, due to electrostatic interaction, results in the release of an ion (H+ or Na+) to the cytoplasm in F-ATPase or periplasm in V-ATPase. Subsequent rotation of the ring disrupts the interaction between the arginine and glutamate residues and permits binding of a new ion from the other side of the membrane via the second half-channel. In the case of F-type ATP synthase, the rotary motion is energetically driven by the electrochemical proton gradient, whereas in the case of V-type ATPases, the rotary motion is driven in the opposite direction by the conformational changes in the V1 domain induced by ATP hydrolysis.
An interesting aspect of the c-ring in F1Fo-ATPases is that the subunit composition differs between different organisms. So far, the number of subunits in c-rings has been found to vary between 10 and 15 (von Ballmoos et al. Reference Von Ballmoos, Cook and Dimroth2008). During each cycle (rotation of 360°), ATP synthase generates three ATP molecules and the number of ions, as well as the strength of the ion motive force required, thus depends on the number of c-subunits. An organism with more subunits will require less ion motive force. There also exist hybrid rotors with a mixture of both F and V-type c subunits in a single ring. In Acetobacterium woodii, the rotor has nine c subunits of F-type each with two TM helices together with one gene-duplicated V-type subunit with four TM helices giving rise to an 11-membered ring (Fritz et al. Reference Fritz, Klyszejko, Morgner, Vonck, Brutschy, Muller, Meier and Muller2008). However, due to the presence of a single charged residue in the V-type subunit only ten ion-binding sites are found. It is not clear how such variations in subunit numbers serve the bacterium and what physiological advantage they provide.
ABC transporters
The superfamily of ABC (ATP-binding cassette) transporters comprise membrane proteins involved in the active uptake of nutrients and others involved in the extrusion of toxic compounds. For example, P-glycoprotein (PGP) is an ABC transporter that plays an important role in the efflux of drugs from cells. The basic architecture of ABC transporters includes two TM and two cytoplasmic domains, which can either be fused into a single polypeptide or be encoded by separate genes. Thus, ABC transporters are either true dimers or have a structurally recognisable twofold axis. The cytoplasmic ATP hydrolysing domains provide the necessary energy for unidirectional transport and the associated TM domains form the substrate translocation pathway. One way of classifying the ABC transporters is based on the direction of transport, i.e. as importers or exporters. Importers are exclusively found in prokaryotes and they rely on additional high-affinity soluble periplasmic proteins to bind the substrate and deliver it to the TM domain. In importers, the TM domain and the ATP binding domain are expressed as separate proteins and then assembled to form a full transporter. ABC exporters are widely distributed from bacteria to mammals and they play a major role in diverse processes such as the extrusion of drugs, transport of lipids or the transport of antigenic/viral peptides for recognition as part of the major histocompatibility complex. Substrates are either taken from the inner leaflet of the membrane bilayer or directly from the cytoplasm (Davidson & Maloney, Reference Davidson and Maloney2007; Locher, Reference Locher2008; Oldham et al. Reference Oldham, Davidson and Chen2008).
The cytoplasmic nucleotide-binding domains (NBDs) contain key motifs that are largely conserved such as the Walker A and B motifs and loops that mediate interaction between the two NBDs or with the TM domains. Perhaps the most important observation from the crystal structures is the head-to-tail cyclic arrangement of the two NBDs, which places the conserved motifs at the interface between the subunits. This results in two ATP molecules being sandwiched at the dimer interface. Such an arrangement allows for positive co-operativity between subunits. Due to the large variety of substrates being transported, the TM domains are widely divergent in both their amino acid sequence and the number and orientation of TM helices. While the exporters known so far all have 12 TM helices, importers contain between 10 and 20 TM helices.
The structures of ABC exporters include the multidrug efflux pumps, Sav1866, P-glycoprotein, and the lipid flippase MsbA (Dawson & Locher, Reference Dawson and Locher2006; Ward et al. Reference Ward, Reyes, Yu, Roth and Chang2007; Aller et al. Reference Aller, Yu, Ward, Weng, Chittaboina, Zhuo, Harrell, Trinh, Zhang, Urbatsch and Chang2009). In the presence of ADP but in absence of a substrate, Sav1866 adopts an outward-facing ATP-bound conformation (Fig. 12b) while P-glycoprotein reveals a substrate bound in an inward facing conformation (Fig. 12a). Both these proteins show an intricate architecture where TM helices adopt a domain-swapped architecture. The interface between TM helices 4 and 5, and TM 10–11 in P-glycoprotein is thought to act as a portal for lateral entry of hydrophobic substrates from the inner leaflet of the membrane. The large cavity observed in P-glycoprotein might explain its ability to transport a broad range of substrates up to a mass of 4000 Daltons and to distinguish stereoisomers (Aller et al. Reference Aller, Yu, Ward, Weng, Chittaboina, Zhuo, Harrell, Trinh, Zhang, Urbatsch and Chang2009). Structures of P-glycoprotein with stereoisomers of cyclic peptides reveal that they bind in different locations and with different stoichiometry (Fig. 12a). The cavity holding the cyclic peptides is largely hydrophobic. Upon ATP hydrolysis and a conformational change to an outward facing conformation, the affinity for substrate is thought to decrease to allow substrate release. In fact, the structure of Sav1866 in its outward facing conformation reveals a hydrophilic cavity close to the inner leaflet with no significant hydrophobic patch (Dawson & Locher, Reference Dawson and Locher2006; Fig. 12b) suggesting how its affinity for a hydrophobic substrate would be reduced.

Fig. 12. ABC (ATP-binding cassette) exporters (a) P-glycoprotein (3G61) and (b) Sav1866 (2HYD). Based on directionality, ABC transporters can be classified as importers or exporters, which use the energy of ATP to import or export substrates. In ABC exporters, the TM (blue and yellow) and nucleotide-binding domains (light blue and light yellow) are expressed either as a single polypeptide, which then associates to form a dimer as in Sav1866 (b) or as one single large polypeptide as in P-glycoprotein (a). The two structures of ABC exporters show an inward facing (a) and outward facing conformation (b). P-glycoprotein crystallised in the presence of substrates, but in the absence of nucleotide, adopts an inward facing conformation with the nucleotide-binding domains spread apart. Substrate (green) binds at an interface between the two halves of the TM domain. In contrast, Sav1866 crystallised in the presence of ADP (orange), reveals an outward facing conformation with the nucleotide-binding domains in close contact. The substrate for prokaryotic Sav1866 is unknown.
Six different importer structures have been solved to date. The simplest of them is a methionine importer (MetNI) with 10 TM helices (Kadaba et al. Reference Kadaba, Kaiser, Johnson, Lee and Rees2008) whereas the molybdate/tungstate transporters from archeae have 12 TM helices (Hollenstein et al. Reference Hollenstein, Frei and Locher2007; Gerber et al. Reference Gerber, Comellas-Bigler, Goetz and Locher2008) and the maltose transporter (MalFGK) from E. coli has 14 TM helices (Oldham et al. Reference Oldham, Khare, Quiocho, Davidson and Chen2007). Although the number of TM helices varies, the orientation of the core helices as defined in the MetNI importer (Fig. 13a) are conserved in the other transporters, indicating a common evolutionary pathway. In contrast, BtuCD (Fig. 13d) and HI1470/71 (Fig. 13e) that import chelated metal have a total of 20 TM helices (Locher et al. Reference Locher, Lee and Rees2002; Hvorup et al. Reference Hvorup, Goetz, Niederer, Hollenstein, Perozo and Locher2007; Pinkett et al. Reference Pinkett, Lee, Lum, Locher and Rees2007) and the relationship in 3D is less obvious. The difference in the number of helices has been attributed to the size of the substrates being transported.

Fig. 13. ABC importers (a) MetNI (3DHW), (b) ModABC (2ONK), (c) MalFGK (2R6G), (d) BtuCD (1L7V) and (e) HIF (2NQ2). Found only in prokaryotes, the domains of importers are encoded and assembled separately (TM domains are in blue and yellow, and the nucleotide-binding domains are in red). Additional regulatory domains can be also found within the NBD such as the C2 domain (cyan) in the MetNI transporter. A high-affinity periplasmic binding protein (green) delivers the substrate to the transporter (ModABC and MalFGK). Based on the TM architecture ABC importers can be subdivided into type I and II importers. Type I importers have a variable number of TM helices (10–14) but the core helices as defined by the minimal MetNI methionine transporter is conserved (a). More commonly observed are 12 TM helices (ModABC and b) while maltose transporter MalFGK has additional peripheral helices (c with additional helices shown in grey). The structure of MetNI (a) without a substrate or nucleotide, and the structure of the molybdate/tungstate transporter (b) from A. fulgidus, with binding protein and substrate (tungstate in magenta) but in the absence of ATP, both reveal inward facing conformations. In MalFGK (c), the presence of ATP (orange) and the binding protein induces an outward facing conformation of the TM domain while the substrate (maltose in magenta) having been released from the binding protein is occluded (Oldham et al. Reference Oldham, Khare, Quiocho, Davidson and Chen2007). Type II importers transport larger substrates such as vitamin B12 and chelated metal. Examples include BtuCD, the vitamin B12 transporter and the homologous HI1470/71, which transports chelated metal. Each monomer has 10 TM helices. BtuCD and HI1470/71 were crystallised in the absence of substrate or nucleotide but reveal outward and inward facing conformations, respectively, probably induced by the detergent environment (Locher et al. Reference Locher, Lee and Rees2002; Pinkett et al. Reference Pinkett, Lee, Lum, Locher and Rees2007; d and e). With all these structures, one can envision a productive cycle, which is described in the main text. However, in the absence of structures from the same protein and considering the probable effects of detergent (BtuCD and HI1470/71), more biochemical and structural data is required to obtain a complete picture of the transport cycle.
The transport cycle of an ABC transporter can be followed based on its nucleotide bound state according to the following hypothetical mechanism. With no nucleotide bound, the NBDs do not interact with one another and the TM domains adopt an inward facing conformation. Binding of ATP results in a closing-up of the NBD domains and the TM domains to create an outward facing conformation, in which an exporter would release the substrate and an importer would accept its substrate from the binding protein. Release of ADP and inorganic phosphate from the NBDs most likely then flips the TM domains to an inward facing conformation where importers release substrate and exporters are ready to accept new substrate.
Although structures for different conformations of the same protein are not yet available, the structures of different transporters provide glimpses of various states of the transport cycle. In the absence of bound nucleotide, ABC transporters adopt an inward facing conformation as seen in multiple structures (Hollenstein et al. Reference Hollenstein, Frei and Locher2007; Pinkett et al. Reference Pinkett, Lee, Lum, Locher and Rees2007; Gerber et al. Reference Gerber, Comellas-Bigler, Goetz and Locher2008; Kadaba et al. Reference Kadaba, Kaiser, Johnson, Lee and Rees2008; Aller et al. Reference Aller, Yu, Ward, Weng, Chittaboina, Zhuo, Harrell, Trinh, Zhang, Urbatsch and Chang2009). Even when the binding protein with its substrate is attached to the transporter, the absence of ATP in the NBDs results in an inward facing conformation (Hollenstein et al. Reference Hollenstein, Frei and Locher2007; Fig. 13b). Upon ATP binding, the TM domain changes its conformation to expose the transport pathway so that it is accessible to the binding protein only from outside. The binding protein then releases the substrate, which diffuses into the transporter. The structure of the MalFGK complex in the presence of maltose and nucleotide probably reflects this intermediate state (Oldham et al. Reference Oldham, Khare, Quiocho, Davidson and Chen2007; Fig. 13c). The BtuCD-F complex has neither the substrate nor the nucleotide bound but is postulated to be an occluded state of the transporter (Hvorup et al. Reference Hvorup, Goetz, Niederer, Hollenstein, Perozo and Locher2007). It is worth mentioning that in the absence of ATP, BtuBC and HI1470/71 adopt outward and inward conformations, respectively, which raises the question of whether the detergent environment might affect the conformational state of the transporter (Locher et al. Reference Locher, Lee and Rees2002; Pinkett et al. Reference Pinkett, Lee, Lum, Locher and Rees2007; Fig. 13d and e).
ABC transporters move substrate against a concentration gradient by directly using the energy from ATP hydrolysis, giving them a more direct link to the primary energy source in comparison with secondary transporters that rely on ion gradients to transport substrate. One interesting question is how cells avoid futile cycles of ATP hydrolysis with no substrate being transported. Some importers have an additional regulatory domain that binds substrate in the cytoplasm thereby keeping the NBD domains far apart and locking the transporter in an inward facing conformation (Gerber et al. Reference Gerber, Comellas-Bigler, Goetz and Locher2008; Kadaba et al. Reference Kadaba, Kaiser, Johnson, Lee and Rees2008). It has also been observed that, in importers, the binding proteins stimulate the rate of ATP hydrolysis in the presence of substrate (Davidson et al. Reference Davidson, Shuman and Nikaido1992). The structure of ModABC from A. fulgidus, which shows the binding protein and substrate still in its inward facing conformation (Hollenstein et al. Reference Hollenstein, Frei and Locher2007; Fig. 13c), probably indicates the requirement of both binding protein and ATP for conformational transition, which might prevent any futile cycle. How the futile cycle is prevented in exporters remains to be addressed.
In eukaryotes, proteins such as the cystic fibrosis transmembrane regulator (CFTR) and TAP transporters that load antigenic peptide on to MHC class I receptors are examples of ABC transporters that have profound physiological and clinical relevance. Structures of prokaryotic transporters have provided a platform to understand the mechanism of ABC transporters. However, the conformational plasticity observed among these transporters and some unique aspects of eukaryotic transporters (for example, CFTR also functions as a chloride channel) calls for many more individual structures in different conformational states to be determined. One interesting study describes how deletion of the cytoplasmic ATP hydrolysing domain in the multidrug transporter, LmrA, results in a functional but proton-dependent transporter (Venter et al. Reference Venter, Shilling, Velamakanni, Balakrishnan and Van Veen2003). Conversely, it is possible that some secondary transporters (discussed below) acquired an ATP hydrolysing domain to become ABC transporters.
Secondary transporters
From the numerous sequenced genomes, the secondary transporters are one of the largest classes of membrane proteins (Paulsen et al. Reference Paulsen, Sliwinski and Saier1998). Secondary transporters carry out a wide variety of functions including transport of many metabolites and ion homeostasis. They are classified into different families based on sequence homology and function. Clustering of transporters according to hydropathy profile analysis distinguishes specific folds and predicts relationships between the transporter families (Lolkema & Slotboom, Reference Lolkema and Slotboom2003), suggesting that they may have a distant evolutionary relationship. Transport of a given substrate can be coupled to H+ ions in one organism or to Na+ ions in another and there is no relation between substrate and ion specificity (Lolkema et al. Reference Lolkema, Speelmans and Konings1994). Whether a specific system is coupled to H+ or Na+ ions is probably related to specific environmental conditions rather than to the optimisation of the catalytic efficiency of secondary transporters.
Like the primary transporters discussed in section 3.1, secondary transporters also undergo large structural changes, but unlike the P-type ATPases or ABC transporters that can be trapped in a particular conformation using ATP analogues, stabilising the secondary transporters in one of the many conformations has proven harder. Hence, well-diffracting crystals of secondary transporters have been relatively difficult to obtain and usually require a careful design of protein construct, the identification of mutants that stabilise one of the many conformations or, more commonly the screening of many different homologues to find one that has a fortuitous predominance of one structure. To date, sixteen structures of individual proteins from thirteen different families of secondary transporter have been determined. Evidence of gene duplication in the evolution of these transporters is clear from these structures (Fig. 14), though there is an enormous variety of gene duplication both in the numbers of TM helices that are duplicated and in their relative orientation, parallel or antiparallel in the membrane.

Fig. 14. Structural gallery of some secondary transporters (a) ATP/ADP carrier (1OKC), (b) LacY (1PV7), (c) EmrE (3B5D) and (d) ClC (1KPL). Secondary transporters make up one of the largest membrane protein families, which is dynamic and diverse. Like many membrane proteins, secondary transporters evolved by gene duplication and fusion. Such repeats are clearly revealed in the recent structures of secondary transporters. The interface between the repeats forms the substrate binding site and translocation pathway. In the ADP/ATP translocase (a), a mitochondrial carrier, the simple repeat is made up of ~100 amino acid helical hairpins that have been triplicated (blue, green and yellow) to give a six-TM helical protein with a pseudo threefold axis relating the hairpins. This structure of ADP/ATP translocase with bound inhibitor carboxyatractyloside (red) shows an outward facing conformation. Duplications of helical bundles are more generally observed resulting in either parallel or antiparallel orientations in the membrane. Lactose permease, LacY (b), a major facilitator superfamily (MFS) transporter has evolved by gene duplication of 2 six helical bundles (blue and yellow) with the same orientation in the membrane. This architecture gives rise to a cavity where substrate (red) binds. Many transporters have their duplicated domains assembled in an antiparallel orientation resulting in two halves wrapped around a common centre bringing amino acids from distinct α-helices together. Occasionally, such duplicated domains are expressed as individual polypeptides and assembled to form an oligomer. EmrE, a small multi-drug transporter (c), is synthesised as two polypeptides (blue and yellow) which are inserted into the membrane in antiparallel orientation, to make an asymmetric homodimer, with inhibitor/substrate tetraphenylphosphonium (red) binding at the monomer-monomer interface. The chloride/proton antiporter, ClC (d), is one such transporter in which the duplicated domains (blue and yellow) are antiparallel in orientation with chloride ions (green spheres) binding at the interface.
The most primitive transporters possibly consisted of multimers of identical domains, presumably pairs of helices as seen in subunit c of the F-type ATPases (Fig. 11). It is logical to imagine that from a simpler structure of multiple identical subunits, evolution favoured a reduction in subunit number by gene fusion together with an increase in the complexity of the sequence, therefore allowing greater specificity or efficiency of a given protein transporter but also functional diversity. The repeats or the duplicated segments can be arranged with parallel or antiparallel topology or be assembled post-translationally with the same or opposite orientation often forming dimers. Thus, one observes a range of manifestations from the simple architecture of a completely membrane embedded transporter such as EmrE (Ubarretxena-Belandia et al. Reference Ubarretxena-Belandia, Baldwin, Schuldiner and Tate2003; Chen et al. Reference Chen, Pornillos, Lieu, Ma, Chen and Chang2007) to a multimeric protein with a large extracellular domain, as in AcrB (Murakami et al. Reference Murakami, Nakashima, Yamashita and Yamaguchi2002) or the intricate architecture of the ClC transporter (Dutzler et al. Reference Dutzler, Campbell, Cadene, Chait and Mackinnon2002). Here, we classify the known structures of secondary transporters based on the arrangement of TM helices.
ADP/ATP translocase and tripartite architecture of mitochondrial carriers
Membrane transport proteins found in the inner mitochondrial membrane are collectively called mitochondrial carriers. All of them are members of the same family with three homologous repeats of about 100 amino acids each resulting in a membrane protein with six TM helices. The presence of triplicated helical hairpins in mitochondrial carriers was first identified from the amino acid sequence of the ADP/ATP translocase (Klingenberg, Reference Klingenberg1981; Saraste & Walker, Reference Saraste and Walker1982). The ADP/ATP translocase is the most abundant and probably the most important carrier in the mitochondrion. It is an electrogenic transporter that exchanges ADP from the cytoplasm with ATP synthesised by F1Fo-ATPase. Despite its natural abundance as the major mitochondrial membrane protein, instability in detergent and susceptibility to proteases resulted in a lack of structural information for more than a quarter of a century (Klingenberg, Reference Klingenberg2008). Fortunately, ADP/ATP translocase in complex with an inhibitor carboxyatractyloside has a higher stability in detergent micelles presumably by trapping the transporter in a single, relatively stable conformation. The structure of the ADP/ATP translocase from bovine mitochondria crystallised in the presence of carboxyatractyloside showed the six TM helical bundle (Fig. 14a) with the helical hairpins related by a pseudo threefold axis and revealed the transporter trapped in an outward facing conformation (Pebay-Peyroula et al. Reference Pebay-Peyroula, Dahout-Gonzalez, Kahn, Trezeguet, Lauquin and Brandolin2003).
Parallel topology
Two duplicated segments typically each containing six TM helices are inserted into the membrane in the same orientation to give a parallel topology, such as in LacY, the lactose transporter (Fig. 14b). The majority of the MFS (major facilitator superfamily – defined because it is the largest superfamily) proteins have this arrangement with a total of 12 TM helices that are divided into two halves related to each other by approximate twofold symmetry. The interface creates a central hydrophilic or hydrophobic cavity that forms the substrate-binding cleft (Hirai et al. Reference Hirai, Heymann, Shi, Sarker, Maloney and Subramaniam2002; Abramson et al. Reference Abramson, Smirnova, Kasho, Verner, Kaback and Iwata2003; Huang et al. Reference Huang, Lemieux, Song, Auer and Wang2003; Yin et al. Reference Yin, He, Szewczyk, Nguyen and Chang2006). AcrB, a proton dependent multidrug transporter belonging to the root nodulation and division family (RND) also has a parallel topology but lacks a cavity at the interface of TM helices. In a similar way to that in the MFS protein family, each monomer of AcrB comprises 12 TM helices and the two halves are related by a twofold axis. Large homologous extracellular domains are inserted within the TM domains between H1 and H2 and between H6 and H7, each of which can be subdivided into portal and TolC docking domains. Hydrophobic substrates that partition into lipids are thought to enter the transporter through the portal domain and be secreted through the outer membrane protein TolC (Murakami et al. Reference Murakami, Nakashima, Yamashita and Yamaguchi2002; Seeger et al. Reference Seeger, Schiefner, Eicher, Verrey, Diedeichs and Pos2006). Alternatively, identical polypeptide chains synthesised separately can also form parallel dimers as observed in the zinc transporter, YiiP, in which each monomer has six TM helices and a cytoplasmic domain (Lu & Fu, Reference Lu and Fu2007). This architecture resembles that of ABC transporters but without the nucleotide-binding domain.
Antiparallel topology
Duplicated segments in secondary transporters are more commonly observed with an antiparallel orientation or opposite topology. This is not exclusive to secondary transporters but is also observed in aquaporins, ammonia channels and the protein translocators, described in sections 2.7, 2.8 and 4.0, respectively. In most transporters such duplication and homology is evident only at the structural level though the MFS duplication was proposed solely from sequence homology in the early 1990s (Marger & Saier, Reference Marger and Saier1993). The structure of the chloride/proton antiporter, ClC, reveals (Fig. 14d) a protein that crosses the membrane 18 times with highly tilted helices that are variable in length (Dutzler et al. Reference Dutzler, Campbell, Cadene, Chait and Mackinnon2002). The N- and C-terminal halves of the protein are in opposite orientations and the two halves wrap around a common centre such that the amino acids from segments of different α-helices are brought together. Similarly, sodium ion-dependent transporters also have an antiparallel topology of duplicated segments. But, in transporters such as the sodium ion/proton antiporter (NhaA; Fig. 15b) or the glutamate transporter (GltPh; Fig. 15a), such duplication is observed only in the helices involved in transport (Fig. 15) but not in the rest of protein (Yernool et al. Reference Yernool, Boudker, Jin and Gouaux2004; Hunte et al. Reference Hunte, Screpanti, Venturi, Rimon, Padan and Michel2005). However, a new structure for GltPh (Reyes et al. Reference Reyes, Ginter and Boudker2009) trapped in an inward-facing conformation by the introduction of a disulphide bridge, shows that, in this state, there is an additional antiparallel topology in the N-terminal region that is not obvious in the outward-facing structure (Yernool et al. Reference Yernool, Boudker, Jin and Gouaux2004).

Fig. 15. Two sodium-dependent transporters (only monomers are shown) (a) GltPh (2NWX) and (b) NhaA (1ZCD). Sodium-dependent transporters have characteristic breaks in their TM helices and these breaks are crucial for ion binding. The architecture of sodium-dependent transporters differs significantly (see text and compare with Fig. 14). The glutamate transporter (GltPh) from Pyrococcus horikoshii and the sodium/proton antiporter (NhaA) from E. coli are two examples of sodium-dependent transporters. GltPh is a homologue of the mammalian excitatory amino acid transporter family. It forms a trimer but each monomer has a separate transport pathway. The monomer of GltPh comprises eight TM helices and two, opposite facing, helical re-entrant hairpins (blue and yellow). Of the eight TM helices, only two (green) associate with the hairpins to form the transport pathway while remaining six helices (grey) form contacts with other monomers. Two sodium ions (violet) and substrate aspartate (red) are found in the transport pathway with residues contributed from the two hairpins and TM helices 7 and 8. Conformational changes in the helical hairpins, TM7 and TM8 are thought to accompany substrate transport. In contrast, NhaA comprises 12 TM helices with two distinctive domains. Six TM helices are involved in transport (blue and yellow) with two discontinuous helices exposing their main chain carbonyls oxygens being responsible for ion co-ordination. The remaining helices (grey) provide a supporting structural role. Unlike in some other transporters (such as those shown in Fig. 14), gene duplications in both GltPh and NhaA are confined only to helices involved in transport and not the entire protein.
Core helices of four distinct sodium-dependent transporters transporting leucine (LeuT; Fig. 16b), galactose (vGLT1; Fig. 16d), hydantoin (Mhp1; Fig. 16a) and betaine (BetP; Fig. 16c) share a similar fold (Yamashita et al. Reference Yamashita, Singh, Kawate, Jin and Gouaux2005; Faham et al. Reference Faham, Watanabe, Besserer, Cascio, Specht, Hirayama, Wright and Abramson2008; Weyand et al. Reference Weyand, Shimamura, Yajima, Suzuki, Mirza, Krusong, Carpenter, Rutherford, Hadden, O'Reilly, Ma, Saidijam, Patching, Hope, Norbertczak, Roach, Iwata, Henderson and Cameron2008; Ressl et al. Reference Ressl, Terwisscha van Scheltinga, Vonrhein, OTT and Ziegler2009) but they show very little if any sequence similarity (Fig. 16). This was noted first by Lolkema & Slotboom (Reference Lolkema and Slotboom2003) using hydropathy profiling prior to any structure determination. The structures of the sodium-dependent transporters show two V-shaped halves (blue and yellow in Fig. 16) that are intertwined with an antiparallel arrangement (Abramson & Wright, Reference Abramson and Wright2009). A common feature observed in six different structures of sodium ion-dependent secondary transporters from different families is the discontinuity in TM helices located in the middle of membrane (Yernool et al. Reference Yernool, Boudker, Jin and Gouaux2004; Hunte et al. Reference Hunte, Screpanti, Venturi, Rimon, Padan and Michel2005; Yamashita et al. Reference Yamashita, Singh, Kawate, Jin and Gouaux2005; Faham et al. Reference Faham, Watanabe, Besserer, Cascio, Specht, Hirayama, Wright and Abramson2008; Weyand et al. Reference Weyand, Shimamura, Yajima, Suzuki, Mirza, Krusong, Carpenter, Rutherford, Hadden, O'Reilly, Ma, Saidijam, Patching, Hope, Norbertczak, Roach, Iwata, Henderson and Cameron2008; Ressl et al. Reference Ressl, Terwisscha van Scheltinga, Vonrhein, OTT and Ziegler2009). Non-helical regions within one or two of these helices expose the main-chain carbonyl oxygens that are involved in sodium ion and substrate binding, also found in the Na+-translocating K-ring of E. hirae discussed in section 3.1.3. A consensus sodium ion-binding motif has been put forward based on these structures and may hold true for all sodium ion-dependent transporters (Boudker et al. Reference Boudker, Ryan, Yernool, Shimamoto and Gouaux2007). A recent addition to the list of membrane proteins with antiparallel topology is the amino acid transporter family of proteins that are sodium independent but have similar architecture to sodium-dependent transporters as well as breaks in the helices in the middle of membrane (Fang et al. Reference Fang, Jayaram, Shane, Kolmakova-Partensky, Wu, Williams, Xiong and Miller2009; Gao et al. Reference Gao, Lu, Zhou, Dang, Sun, Li, Wang and Shi2009; Shaffer et al. Reference Shaffer, Goehring, Shankaranarayanan and Gouaux2009).

Fig. 16. Different conformational states of sodium dependent secondary transporters: (a) Mhp1, hydantoin transporter (2JLN), (b) LeuT, leucine transporter (2A65), (c) BetP, betaine transporter (2W8a) and (d) vGlt1, galactose transporter (3DH4). Different conformations have been observed in structures of the sodium-dependent transporters Mhp1, LeuT, BetP and vGlt1. Although, these transporters show little sequence homology a core structure of 10 TM helices (blue and yellow) involved in substrate translocation share a recognisably similar fold. Additional helices (grey) might have evolved for specific needs or modulation. The structure of Mhp1 with no substrate reveals an outward facing conformation, while LeuT with leucine (red) and two sodium ions (purple) reveals a substrate-occluded but outward facing state. BetP with substrate bound (red) probably represents an intermediate occluded state with both gates closed. In contrast, vGlt1in the presence of galactose (red) and a sodium ion (purple) reveals an inward facing conformation. These structures indicate the existence of multiple states, but it is not clear whether a particular transporter has its own preferred state to crystallise or the presence of detergent, substrate or crystallisation conditions modulates the series of states in the transport cycle. Discontinuous helices in all four transporters are involved in substrate or sodium ion binding. Conformational changes are thought to occur in the discontinuous helices accompanied by changes in extracellular and intracellular gates resulting in substrate translocation.
In other families of secondary transporters, single polypeptide chains typically of about 100 amino acids are inserted into the membrane to form dimers in which the two identical polypeptide chains have opposite orientations. EmrE, a member of the small multidrug resistance family, is a membrane protein of this type (Fig. 14d). It comprises four TM helices from each monomer, which associate to form an eight-helix antiparallel dimer resulting in a central cavity that forms the translocation pathway (Ubarretxena-Belandia et al. Reference Ubarretxena-Belandia, Baldwin, Schuldiner and Tate2003; Chen et al. Reference Chen, Pornillos, Lieu, Ma, Chen and Chang2007). Interestingly, the two chemically identical polypeptide chains have different conformations, so that the whole structure is an asymmetric homodimer.
Mechanism of secondary transporters
Although monomers are often sufficient for transport, dimers or trimers are frequently observed. Formation of such oligomers could aid in interactions with other proteins or could allow co-operativity between subunits (Murakami et al. Reference Murakami, Nakashima, Yamashita, Matsumoto and Yamaguchi2006; Seeger et al. Reference Seeger, Schiefner, Eicher, Verrey, Diedeichs and Pos2006), or perhaps could simply increase stability. In a given family the quaternary structure and architecture of membrane spanning domains are often mutually conserved (Hirai et al. Reference Hirai, Heymann, Shi, Sarker, Maloney and Subramaniam2002; Abramson et al. Reference Abramson, Smirnova, Kasho, Verner, Kaback and Iwata2003; Huang et al. Reference Huang, Lemieux, Song, Auer and Wang2003; Yin et al. Reference Yin, He, Szewczyk, Nguyen and Chang2006). In some transporters, the oligomeric state has been reported to differ dependent on whether the protein is in a lipid bilayer or in a detergent micelle. The ADP/ATP translocase and the NhaA antiporter are two such examples where dimers are observed in native membranes or in 2D crystals (Williams, Reference Williams2000; Klingenberg, Reference Klingenberg2008) but structures determined in detergents reveal them as monomers (Pebay-Peyroula et al. Reference Pebay-Peyroula, Dahout-Gonzalez, Kahn, Trezeguet, Lauquin and Brandolin2003; Hunte et al. Reference Hunte, Screpanti, Venturi, Rimon, Padan and Michel2005). This is probably due to delipidation during purification. Weak interactions between monomers involving protein:lipid:protein interactions are disrupted by detergent (Nury et al. Reference Nury, Dahout-Gonzalez, Trezeguet, Lauquin, Brandolin and Pebay-Peyroula2005; Appel et al. Reference Appel, Hizlan, Vinothkumar, Ziegler and Kuhlbrandt2009).
A desirable goal, not yet attained, is to determine multiple structures representing the complete transport cycle of a single transporter. While the alternating access model (Jardetzky, Reference Jardetzky1966) predicts the existence of at least two conformations of a transporter in vivo, a crystal structure usually reveals the static state of only one of these conformations. In fact a number of different conformational states have been observed in the published structures from different proteins belonging to the same family. Structures of lac permease and the glycerol phosphate transporter from the MFS family reveal an inward facing conformation (Abramson et al. Reference Abramson, Smirnova, Kasho, Verner, Kaback and Iwata2003; Huang et al. Reference Huang, Lemieux, Song, Auer and Wang2003), while EmrD and a low-resolution EM map of OxlT from the same family probably reveal a substrate-occluded state (Hirai et al. Reference Hirai, Heymann, Shi, Sarker, Maloney and Subramaniam2002; Yin et al. Reference Yin, He, Szewczyk, Nguyen and Chang2006).
Four structures of sodium-dependent transporters provide another example of conformational diversity where two of these transporters, Mhp1 and LeuT, reveal an outward facing conformation in the absence or presence of substrate (Yamashita et al. Reference Yamashita, Singh, Kawate, Jin and Gouaux2005; Weyand et al. Reference Weyand, Shimamura, Yajima, Suzuki, Mirza, Krusong, Carpenter, Rutherford, Hadden, O'Reilly, Ma, Saidijam, Patching, Hope, Norbertczak, Roach, Iwata, Henderson and Cameron2008; Fig. 16a and b) while vGLT1 shows substrate bound in an inward facing conformation (Faham et al. Reference Faham, Watanabe, Besserer, Cascio, Specht, Hirayama, Wright and Abramson2008; Fig. 16d). The structure of BetP with substrate bound has been proposed to be an intermediate state (Ressl et al. Reference Ressl, Terwisscha van Scheltinga, Vonrhein, OTT and Ziegler2009; Fig. 16c). Comparison of these four structures provides a plausible sequence for the mechanism of transport. Conformational change is thought to occur by rotation of the cytoplasmic part of the discontinuous helices accompanied by changes in extracellular or intracellular gates that open and close the pathway (Faham et al. Reference Faham, Watanabe, Besserer, Cascio, Specht, Hirayama, Wright and Abramson2008). Apart from their role in substrate binding, such breaks in helices must play an important role in the transport cycle. This is in contrast to the MFS family of transporters that use a shared binding site at the interface of two helical halves in which the transport cycle probably involves more global changes in TM helices, similar to that found in the ABC transporters.
Is it possible to observe multiple conformations in the same structure? AcrB, the multi-drug exporter crystallises as a symmetric as well as an asymmetric trimer. In the asymmetric trimer, only one monomer has the drug bound and comparison of monomers reveals three distinct states, proposed to be tight, open and loose, giving rise to the proposal of a possible cyclical mechanism in which functional states rotate round the trimer (Murakami et al. Reference Murakami, Nakashima, Yamashita, Matsumoto and Yamaguchi2006; Seeger et al. Reference Seeger, Schiefner, Eicher, Verrey, Diedeichs and Pos2006).
Secondary transporters in humans are implicated in many physiological processes and are important drug targets. Except for the mitochondrial ADP/ATP translocase, other secondary transporter structures that have been determined so far have come exclusively from prokaryotic sources, raising the question of whether they are truly relevant to the architecture of mammalian proteins. LeuT from Aquifex aeolicus, a homologue of neurotransmitter transporters, provides an excellent example of how structures from prokaryotic homologues can be useful to extend our understanding of transporters from higher eukaryotes. The hallmarks of the neurotransmitter transporter family in eukaryotes include a dependence on sodium and chloride ions for transport as well as inhibition by tricyclic antidepressants (TCAs). Structures of LeuT with TCA bound show that TCAs probably inhibit LeuT by binding to a secondary site near the surface of the protein that is distinct from the substrate binding site in the centre of membrane (Singh et al. Reference Singh, Yamashita and Gouaux2007; Zhou et al. Reference Zhou, Zhen, Karpowich, Goetz, Law, Reith and Wang2007). Binding of TCA is thought to stabilise the extracellular gate in a closed conformation thereby preventing the release of substrate. A recent study shows that octyl glucoside, the detergent used for crystallising LeuT, occupies the same site as TCA and can also inhibit LeuT, thus prompting a suggestion that all structures of LeuT determined so far are blocked in the outward facing conformation (Quick et al. Reference Quick, Winther, Shi, Nissen, Weinstein and Javitch2009). The substrate of LeuT, leucine, also binds to this secondary site, which has been proposed to be essential for driving the transporter into a cytoplasmic-facing conformation that subsequently releases substrate. These observations could explain some of the characteristics of eukaryotic neurotransmitter transporters.
Secondary transporters have a wide range of transport rates, varying from about 10 molecules per second to 104 ions per second in some ion transporters. Increasingly, it is becoming clear that at least some secondary transporters, such as the human serotonin transporter SERT (Li et al. Reference Li, Farley and Lester2002), also exhibit channel-like properties. In fact, the chloride/proton antiporter was initially classified as a channel but subsequent electrophysiological analysis revealed it to be an antiporter with a very high transport rate (Accardi & Miller, Reference Accardi and Miller2004). Structural explanations, involving small conformational changes, of how such high transport rates can be achieved are starting to emerge. It is likely that the different topological arrangements of gene-duplicated TM helix domains (parallel or antiparallel) led to different mechanisms of conformational change and associated differences in the transport rate. The relatively high number of transporter genes found in sequenced genomes is likely to represent a relatively small number of unique structures and translocation mechanisms, but multiple structures from a given family are still needed.
Protein translocators
In bacteria, membrane proteins are inserted into the cytoplasmic or periplasmic membranes either post-translationally or co-translationally and both routes also occur for the secretion of soluble proteins. Both mechanisms make use of a protein translocation channel, the SecY complex, consisting in bacteria of a 1:1:1 trimer of SecY, SecE and SecG. During co-translational protein export, the SecY complex binds directly to the ribosome and the nascent polypeptide chain exiting from the ribosome drives membrane protein insertion. During post-translational export, the SecY complex binds to SecA, which drives export using the energy from ATP hydrolysis to push the polypeptide through the SecY channel (Zimmer et al. Reference Zimmer, Nam and Rapoport2008). In eukaryotes, the homologous complex is the Sec61αβγ complex. Although there has been some controversy about whether the complex functions as a monomer or higher order oligomer and the crystal structures show only monomeric complexes (Fig. 17), the best current evidence (Menetret et al. Reference Menetret, Schaletzky, Clemons, Osborne, Skanland, Denison, Gygi, Kirkpatrick, Park, Ludtke, Rapoport and Akey2007; Menetret et al. Reference Menetret, Hegde, Aguiar, Gygi, Park, Rapoport and Akey2008; Zimmer et al. Reference Zimmer, Nam and Rapoport2008) indicates the monomer is the functional unit.

Fig. 17. Protein translocation channels: (a) SecYEβ (1RHZ) and (b) SecA–SecYEG (3DIN). Monomeric SecYEβ in M. jannaschii forms the minimal protein translocation unit. The channel forming SecY is made up of 10 TM helices that can be divided into two equal halves (blue and yellow) related by a pseudo twofold symmetry axis parallel to the plane of the membrane. Like many other membrane proteins, the two halves thus have an antiparallel orientation. SecE has a short amphipathic helix and a very long highly tilted TM helix (green) and forms extensive contact with SecY. The β-subunit (purple) is oriented perpendicular to the membrane and lies just outside SecY. Together SecE and Secβ are thought to provide structural integrity to SecY during protein translocation. A small helix (red) acts as a plug to seal the channel when no protein is translocated. A complex between SecA and SecYEG of T. maritima was obtained in the presence of ADP and BeCl2. SecG of T. maritima (equivalent to the β-subunit of M. jannaschii) has an additional TM helix (purple). The five domains of SecA are coloured individually. The two nucleotide-binding domains are coloured wheat and grey with a molecule of ADP shown in black. The polypeptide-crosslinking and helix wing domains are shown in aquamarine and orange, respectively. The helical scaffolding domain (brown) of SecA makes contact with the C-terminal domain of SecY. A comparison of the two structures reveals movement of three TM helices in SecY, which opens up the central channel and is accompanied by displacement of the plug helix (red). Channel opening allows the polypeptide to be secreted either to the periplasm or laterally into the membrane.
SecY, the channel forming subunit has 10 TM helices that are arranged as 2 five-helix bundles; TM1-TM5 in one half of SecY and TM6-TM10 in the other half are related by a pseudo twofold symmetry axis parallel to the membrane (Fig. 17a). Although some of the transporters with antiparallel topology also have 2 five-helix inverted repeats, the detailed arrangement of helices in SecY is quite different. There is an hourglass-shaped pore between the two SecY domains with funnels to the cytoplasmic and external sides of the membrane. Subunits SecG and SecE are located on the periphery of the complex, perhaps to provide structural stability to SecY, which is believed to undergo fairly large structural changes during protein export. SecG has a single TM helix in Methanococcus jannaschii (Van den Berg et al. Reference Van den Berg, Clemons, Collinson, Modis, Hartmann, Harrison and Rapoport2004) but there are two TM helices in SecG from Thermotoga maritima (Tsukazaki et al. Reference Tsukazaki, Mori, Fukai, Ishitani, Mori, Dohmae, Perederina, Sugita, Vassylyev, Ito and Nureki2008; Zimmer et al. Reference Zimmer, Nam and Rapoport2008) with the C-terminal helix having the same location as the single TM helix in M. jannaschii. SecE is highly tilted and makes extensive contacts with TM1, TM5, TM6 and TM10 of SecY. In the absence of SecA or a ribosome, the cytoplasmic funnel is empty and has a diameter of about 25 Å, whereas the extracellular funnel is occupied by a short plug helix TM2a.
The ATP driven SecA has five domains: two N-terminal RecA-like domains that combine to bind nucleotide, a polypeptide-cross-linking domain (PPXD), a helical wing domain and the helical scaffold domain (HSD). In the protein translocation field, there is a lot of interest in understanding the nature of the structural changes in SecA when a peptide binds, followed by how SecA binds to SecY and how SecA and SecY work together during transport. A complex of SecA with SecYEG obtained in presence of ADP and BeCl2 is shown in Fig. 17b viewed parallel to the membrane. The main contact between SecA and SecYEG is made through the HSD. Comparison of the structure of SecYEG alone (van den Berg et al. Reference Van den Berg, Clemons, Collinson, Modis, Hartmann, Harrison and Rapoport2004; Fig. 17a) with that in complex with SecA (Zimmer et al. Reference Zimmer, Nam and Rapoport2008; Fig. 17b) shows that there is an outward movement of the C-terminal domain of SecY (yellow in Fig. 17a) when SecA binds. Movements of three helices, specifically TM7, TM8 and TM9, open up the centre of the channel. This allows a two-helix finger, corresponding to the two shorter helices of the three helices that make up the helical scaffold domain HSD of SecA, to insert into the cytoplasmic funnel of SecY. At the same time, TM2a, the plug helix in the external funnel of SecY, moves to a more external position. These observations have allowed Zimmer et al. (Reference Zimmer, Nam and Rapoport2008) to propose a very plausible conceptual model for how SecA-mediated transport pushes the polypeptide through the SecY channel: ATP is hydrolysed and SecA alternately clamps and releases the polypeptide as it is pushed through the channel.
Presumably the co-translational mechanism of protein export by direct interaction with the ribosome involves a similar mechanism (Menetret et al. Reference Menetret, Hegde, Aguiar, Gygi, Park, Rapoport and Akey2008) with the ribosome replacing SecA to provide the push. Proteins destined for the membrane, most of which are SecA independent, are thought to be released through a lateral gap that opens up between TM7 and TM2b. In summary, regular α-helices in SecY undergo relatively large movements of up to 10 Å laterally in the membrane to provide the flexibility needed for protein export, while ensuring that the channel is closed and does not leak when not involved in protein transport.
Electron transport chains
Energy in cells is obtained either from oxidation of molecules from food or from capture of light energy through photosynthesis. Each pathway couples a sequence of electron transfer reactions to synthesis of ATP, from ADP and Pi, via the intermediate energy store of an electrochemical gradient of hydrogen ions across the membranes of bacteria, chloroplasts or mitochondria. The electron transfer and associated proton transfer steps occur directionally across the membrane to produce the electrochemical gradient. Peter Mitchell in 1960 first proposed the overall mechanism, the chemiosmotic theory, for which he was awarded the 1978 Nobel Prize in Chemistry (Mitchell, Reference Mitchell1979). The protein:chlorophyll complexes in photosynthesis that are involved in the primary capture of light energy upstream of the electron transport chain are discussed later.
In this section, we discuss the structures of the membrane protein complexes that make up the electron transport chain. This chain carries out the sequence of reactions in which the high potential energy of electrons from NADH drops down through a sequence of intermediates (ubiquinone and cytochrome c) ending with the reduction of oxygen to produce water. There are five complexes. Complex I is NADH:ubiquinone reductase, which releases the lipophilic reduced ubiquinol into the membrane. Complex III then carries out the ubiquinol:cytochrome c reductase reaction. Finally, in complex IV, cytochrome c reduces molecular oxygen to water in the cytochrome c oxidase reaction transferring four protons to the site of oxygen reduction, and pumping four more across the membrane. These three membrane protein complexes each couple an electron transfer step to the transfer of protons across the membrane to create the electrochemical membrane potential, which is then used by complex V, the ATP synthase already discussed in section 3.1.3, to synthesise ATP. Complex II, succinate:ubiquinone reductase, is the fifth member of the membrane bound electron transport chain; it takes in electrons tangentially from succinate and reduces ubiquinone to ubiquinol, but this transfer, unlike at other complexes in the chain, is not coupled to proton transfer since the free energy difference is too small.
Much of the current research in this field is directed towards understanding the mechanism of coupling between electron transfer and proton transfer in complexes I, III and IV, but in this review, we focus only on the general architecture of the proteins and the associated arrangement of the redox centres through which the electrons pass. In addition to these three integral membrane protein complexes, mitochondria also have acyl-CoA dehydrogenases and electron-transfer flavoproteins to transfer electrons from fatty acid β-oxidation to the coenzyme Q pool. These are membrane bound rather than integral membrane proteins and are mentioned briefly in section 8.5. In prokaryotes, alternative electron transport pathways exist that allow an organism to use terminal electron acceptors other than molecular oxygen, which include nitrogen (Jormakka et al. Reference Jormakka, Tornroth, Byrne and Iwata2002; Bertero et al. Reference Bertero, Rothery, Palak, Hou, Lim, Blasco, Weiner and Strynadka2003) and sulphur (Jormakka et al. Reference Jormakka, Yokoyama, Yano, Tamakoshi, Akimoto, Shimamura, Curmi and Iwata2008) but also elements like selenium, iron and manganese (Jormakka et al. Reference Jormakka, Byrne and Iwata2003). Detailed description of alternative electron transport pathway is beyond the scope of this review, in this section we focus only on the electron transport chain with molecular oxygen as the terminal electron acceptor.
Each electron transport complex is a multi-subunit protein with electron-carrying cofactors including flavins, iron–sulphur clusters, haems and copper ions. These redox cofactors are bound either to membrane embedded TM helical domains within each complex or to hydrophilic domains that protrude on the cytoplasmic or periplasmic sides of the membrane. Structures of complexes II, III and IV are shown in Fig. 18.

Fig. 18. Electron transport chain: (a) complex II (1ZOY), (b) complex III (2FYU) and (c and d) complex IV (1V54). (a) Complex II, functional as a monomer as illustrated, shows four subunits of which two are membrane spanning each with three TM helices (blue and yellow), together with five cofactors, of which only the haem b (red) and the quinone binding site (not shown) are located at the level of the membrane. FAD (orange) and three iron–sulphur clusters (brown) are also shown. (b) Complex III, showing only one half of the dimer that has the TM helix of the ISP-Rieske subunit (cyan) domain swapped with its TM helix (protruding on the left side of the molecule) interacting more closely with the other monomer. There are eight subunits in the bovine mitochondrial enzyme, of which the cytochrome b subunit is most important (blue). It contains eight TM helices in two bundles, of five and three helices. The five-helix bundle forms the binding site for the two b-type haems (red) that are aligned vertically above one another with each haem iron being coordinated by two histidine side chains that are 14 residues apart (four turns) on helices B and D. The other three helices of cytochrome b and the remaining five single TM helices from other subunits have peripheral roles (grey). Cytochrome c1 (yellow) with its c-type haem (brown) is located in the intramembrane space. The cytoplasmic/matrix subunits (grey) are also not directly involved in electron transport. (c) Complex IV, showing a side view of only the two functionally important subunits I and II (blue and yellow), together with the three metal centres: the CuA two-copper site in subunit II (brown spheres), and the sites for haem a (red) and binuclear haem a3/CuB (red and green) in subunit I. (d) Top view of complete monomer of bovine mitochondrial complex IV, showing all 28 TM helices in cross-section. Only the 12 helices (blue) of subunit I and the two helices of subunit II (yellow) are directly involved in electron transport and proton pumping. These 12 TM helices in subunit I are related by an approximate threefold axis, easily visible in the picture with haem a (red) being located in one sector, haem a3/CuB (red and green) being located in the second sector and proton channel D being located in the third.
Complex I – NADH:ubiquinone reductase
At this time, there is no high resolution structure of the membrane-embedded part of complex I, though there is a good 3·3-Å resolution structure (Sazanov & Hinchliffe, Reference Sazanov and Hinchliffe2006) for the large cytoplasmic hydrophilic domain of complex I from Thermus thermophilus, showing eight protein subunits, plus cofactors FMN and a chain of nine iron–sulphur clusters. Indeed all the known redox centres of complex I are found in this hydrophilic domain. At least seven more protein subunits in bacteria make up the membrane domain. There are low-resolution structures of the whole complex from both bacteria and mitochondria determined by electron microscopy (Grigorieff, Reference Grigorieff1998; Guenebaut et al. Reference Guenebaut, Schlitt, Weiss, Leonard and Friedrich1998). Sequence homology suggests that the large hydrophobic subunits of complex I might be homologous to the Na+/H+ antiporter family discussed in section 3.2.3 (Berrisford et al. Reference Berrisford, Thompson and Sazanov2008) and therefore designed to drive proton transfer via protein conformational changes. However, there is no real clue yet as to the nature of the presumed conformational changes that might couple electron transport in the hydrophilic domain to proton transfer/pumping in the membrane domain.
Complex II – succinate:ubiquinone reductase
Complex II, with four protein subunits (Fig. 18a), funnels electrons from the succinate oxidation site at the FAD in the middle of the cytoplasmic hydrophilic domain (equivalent to the matrix side of the inner membrane in mitochondria) through a 40-Å path made up of 3 distinct iron–sulphur clusters ending up with reduction of ubiquinone in the membrane (Yankovskaya et al. Reference Yankovskaya, Horsefield, Tornroth, Luna-Chavez, Miyoshi, Leger, Byrne, Cecchini and Iwata2003; Sun et al. Reference Sun, Huo, Zhai, Wang, Xu, Su, Bartlam and Rao2005). The haem b site lies within the TM helical bundle whereas the quinone binding site is on the lipid facing surface of the TM bundle. The two hydrophobic membrane spanning subunits (CybL (large) and CybS (small) in pig) each have three TM helices that surround the single haem b, making a simple ring of six TM helices related by an approximate twofold axis and with the helices, from amino- to carboxy-terminus, running clockwise viewed from the cytoplasmic side. Intriguingly, the path of the electron from FAD to quinone does not appear to pass through the haem, so it is believed that the role of the haem may be to create an electron sink that minimises production of reactive oxygen species (Tran et al. Reference Tran, Rothery, Maklashina, Cecchini and Weiner2007). The N-termini of the membrane subunits are inside (i.e. exposed to the cytoplasm or matrix, respectively) and the C-termini outside the bacterial or mitochondrial inner membrane. Thus, the membrane domain of complex II is a classical six-helix transmembrane bundle with relatively straight helices. The membrane spanning subunits are both smaller than the two cytoplasmic (or matrix) subunits that bind flavin and the iron–sulphur clusters. Other than the hydrophobicity of the substrate ubiquinone, there is no other reason why complex II needs to be a membrane protein, so perhaps this is why the membrane domain is so simple. The related membrane enzyme fumarate reductase shows similar structures but with variations in the arrangement of TM helices and the number of haem groups (Lancaster et al. Reference Lancaster, Kroger, Auer and Michel1999).
Complex III – ubiquinol:cytochrome c reductase
The number of protein subunits in complex III varies from 3 in Paracoccus denitrificans or Rhodopseudomonas sphaeroides to 9 or 10 in yeast (Hunte et al. Reference Hunte, Koepke, Lange, Rossmanith and Michel2000) and 11 in bovine mitochondria (Iwata et al. Reference Iwata, Lee, Okada, Lee, Iwata, Rasmussen, Link, Ramaswamy and Jap1998; Esser et al. Reference Esser, Gong, Yang, Yu, Yu and Xia2006). Only three subunits, common to all species, have key roles and each contains a unique cofactor. The large cytochrome b subunit has two b-type haems embedded within its eight TM helix bundle (Fig. 18b). The globular cytochrome c1 subunit, with its c-type haem covalently thioether-linked to cysteine amino acids, is located in the periplasmic (or intermembrane in mitochondria) space with a single membrane-anchoring TM helix and finally, the Rieske iron–sulphur cluster (2Fe-2S; ISP), located adjacent to cytochrome c1, also has a single membrane-anchoring TM helix. Overall, complex III is a multi-subunit dimer with a small degree of domain swapping in that the hydrophilic iron–sulphur domain of one ISP interacts with one cytochrome b monomer whereas its membrane-anchoring helix interacts with the other cytochrome b monomer. The ISP redox centre and possibly also cytochrome c1 are fairly mobile moving by up to 15 Å between different crystal structures, reflecting their role in electron transfer by binding to, reducing and releasing successive cytochrome c molecules. The eight non-conserved subunits are believed to have roles in assembling, stabilising, modulating, regulating or increasing the efficiency of the activity. The three subunits common to all species, not surprisingly, have the greatest structural conservation and are the key to understanding the function.
The eight TM helices in cytochrome b are arranged as two bundles, a five-helix N-terminal bundle located near the dimer twofold axis that binds the two haems, and a more distant three-helix C-terminal bundle. The four histidine side chains that are the axial ligands to the two b-type haems are located 14 residues (four turns) apart on helices B and D facing the centre of the protein. Helices A, B, C and D are arranged clockwise around the haems viewed from the cytoplasm/matrix side, as in complex II. The two quinone binding sites are accessible from hydrophobic clefts within the membrane at the interface between the two cytochrome b subunits, with the Qo site being close to the low potential haem bL on the intermembrane side and Qi being close to the high potential haem BH on the matrix side. In the overall Q-cycle reaction, one of the two electrons from quinol in the Qo site is transferred via ISP and cytochrome c1 to reduce cytochrome c, whereas the other reduces the Qi quinone. The protons that are an intrinsic part of the redox reaction are released from quinol in the Qo site and taken up by quinone at the Qi site on the other side of the membrane, thus converting the electron transfer energy into an electrochemical proton gradient.
There are two very large cytoplasmic/matrix subunits, known as the core 1 and core 2 subunits in the bovine mitochondrial enzyme. These are conserved in eukaryotes but absent in bacteria. Their amino acid sequences show clear homology to matrix processing peptidases (MPP), which are zinc metalloproteases. The bovine bc1 complex has only weak protease activity and in yeast there is no protease activity at all since the amino acid side chains that are normally the zinc ligands have mutated so that no zinc is bound. The role of core 1 and core 2 thus appears to be that of a structurally important relic of some earlier function. There are three other TM helices from three smaller peripheral subunits within each monomer of the bc1 complex, whose role is less important.
A related complex, cytochrome b6f which is a plastoquinol:plastocyanin reductase, is found in chloroplasts. It catalyses the redox step linking photosystems I and II, whose structures are described in section 6. The four TM helices and the 2 b-type haems in cytochrome b6 are homologous to the four N-terminal helices and the two haems of cytochrome b (Kurisu et al. Reference Kurisu, Zhang, Smith and Cramer2003; Stroebel et al. Reference Stroebel, Choquet, Popot and Picot2003). The three TM helices of subunit IV of the b6f complex are located in the same region as the four C-terminal TM helices of cytochrome b. Cytochrome f and c1 are functionally the same but structurally quite different. The redox roles of ubiquinol and cytochrome c in the bc1 complex are played by plastoquinol and plastocyanin in b6f. In cytochrome b6, there is also an extra haem on the stromal side, equivalent to the matrix side in mitochondria, as well as a chlorophyll and a carotenoid molecule both thought to play structural, non-redox roles (Kurisu et al. Reference Kurisu, Zhang, Smith and Cramer2003). Finally there are also four smaller peripheral subunits in the b6f complex, each with a single TM helix, occupying positions unrelated to the extra single-spanning subunits in the bc1 complex.
Complex IV – cytochrome c oxidase (aa3)
Cytochrome oxidase consists of three to five protein subunits in bacteria and up to 13 in bovine mitochondria. Of these, only subunits I and II (Fig. 18c) are absolutely essential for the oxidation of cytochrome c and the reduction of oxygen to water. The other subunits presumably help assembly, regulation or efficiency. As the terminal oxidase, cytochrome oxidase is the final step in oxidation of substrate by molecular oxygen. Even minor improvements in the efficiency of energy conversion will have been locked in by evolution. Thus, the overall complexity of the structure masks the intrinsic simplicity of the reaction. The overall reaction is
The enzyme stores the first two electrons in the absence of oxygen on haem a3 iron and CuB. After this two electron reduction, cytochrome oxidase reacts rapidly with oxygen to split the O=O bond, assisted by proton and electron transfer from the covalent active-site tyrosine, with the third and fourth electrons arriving via CuA and haem a. The four electron reduction of oxygen to produce two water molecules is accompanied by uptake of four chemical protons to maintain electroneutrality, but as first demonstrated by Wikstrom in 1977, each electron transfer is also accompanied by the pumping of up to one proton across the membrane thus doubling the electrochemical gradient created by the electron and chemical proton transfer reactions and making greater use of the redox energy drop between cytochrome c and oxygen. The location of the proton pumping channel, the nature of the proton pumping group and how any structural change might cause pK changes and control accessibility is still controversial, though there is a steady flow of publications from several groups addressing the problem. The most convincing current view (Brzezinski & Gennis, Reference Brzezinski and Gennis2008) is that the energy from purely electrostatic interactions between the electrons and protons as they accumulate at the binuclear a3/CuB site, before and after oxygen binding, drives the proton pumping during each cycle of electron transfer from CuA to haem a to the a3/CuB binuclear centre.
The 1·8-Å structure of the 13 subunit bovine cytochrome oxidase (Tsukihara et al. Reference Tsukihara, Aoyama, Yamashita, Tomizaki, Yamaguchi, Shinzawa-Itoh, Nakashima, Yaono and Yoshikawa1996; Yoshikawa et al. Reference Yoshikawa, Shinzawa-Itoh, Nakashima, Yaono, Yamashita, Inoue, Yao, Fei, Libeu, Mizushima, Yamaguchi, Tomizaki and Tsukihara1998; Tsukihara et al. Reference Tsukihara, Shimokata, Katayama, Shimada, Muramoto, Aoyama, Mochizuki, Shinzawa-Itoh, Yamashita, Yao, Ishimura and Yoshikawa2003; Aoyama et al. Reference Aoyama, Muramoto, Shinzawa-Itoh, Hirata, Yamashita, Tsukihara, Ogura and Yoshikawa2009) in the fully oxidised state, shown in Fig. 18c and d, shows all the features present in complexes with fewer subunits from other species, though key mechanistic insights have also been obtained from structures of bacterial cytochrome oxidases especially the minimal, two subunit enzymes from P. denitrificans (Ostermeier et al. Reference Ostermeier, Harrenga, Eermler and Michel1997) and R. sphaeroides (Qin et al. Reference Qin, Hiser, Mulichak, Garavito and Ferguson-Miller2006). Analysis of uncoupled mutants has also helped to understand the mechanism of proton pumping.
The key features of the structure are:
1. Although the monomer is the functional unit, the bovine mitochondrial enzyme exists as a dimer in vivo and in the crystals.
2. Subunit I shows an internal approximate threefold axis relating three ‘pores’ that house haem a, the haem a3/CuB bimetal centre and one of the proton channels that have been defined by analysing mutants.
3. Subunit II contains the CuA sites (two copper atoms) and two TM helices that act as an anchor.
4. Subunit III has seven more helices with a subsidiary role, since the two subunit enzyme has full activity.
5. There are further subsidiary subunits with single TM helices, consisting of the single subunit SuIV in bacteria or seven additional subunits in bovine mitochondria.
6. There is a unique spontaneous covalent bond formed between H240-Nε and Y244-Cε in the bovine enzyme, conserved in all family A and B oxygen reductases, which reduces the pK of the tyrosine hydroxyl. H240 is one of the CuB ligands and the tyrosine hydroxyl group points directly at the oxygen binding site, so it is believed that it must have a role in stabilising at least one of the intermediates in the four electron reaction, possibly even as a free radical.
7. There are two proton channels, named D and K from the mutations that originally identified them, but how they function is still under investigation.
8. The O2 binding site bridges between the haem a3 iron atom and CuB, and is otherwise occupied by hydroxyl and water.
9. The sequence of electron transfers from binuclear CuA to haem a to haem a3/CuB is likely to occur through a series of H-bonded groups.
The overall function of cytochrome oxidase is of great interest because of its biological importance, its chemical complexity, and the tightness of the coupling between electron transfer, charge separation and proton pumping. Although there are small changes in structure between the fully oxidised and fully reduced structures (Yoshikawa et al. Reference Yoshikawa, Shinzawa-Itoh, Nakashima, Yaono, Yamashita, Inoue, Yao, Fei, Libeu, Mizushima, Yamaguchi, Tomizaki and Tsukihara1998), which may be part of a proton pump gating mechanism in bovine mitochondria, the overall structure is remarkably rigid, acting as a framework for electron and proton transfers that are mainly controlled by electrostatics. This is in marked contrast to the very much larger structural changes that characterise the primary and secondary transporters that transport or pump much larger substrates. In fact, the structures of the transmembrane helix bundles of all three electron transfer complexes discussed in this section are relatively rigid and immobile.
Photosynthesis
In this section, we consider only those aspects of photosynthesis that involve membrane proteins or membrane protein complexes whose structure is known in enough detail to resolve the polypeptide chain and cofactor or pigment structure. The key structures are of two kinds: those that absorb incident light and those that convert the light energy into chemical or electrochemical energy by charge separation and electron transfer.
Primary light absorption occurs in the light-harvesting (LH) complexes (LHCs), which then funnel light energy, by Förster energy transfer, directly to the reaction centres jumping over distances between neighbouring chromophores of 10–25 Å. Primary charge separation occurs within the reaction centres (RC), leading to creation of a membrane potential, or reduction of a cofactor such as a quinone, or both. This intermediate energy store is subsequently converted into ATP through the electron transport chain and oxidative phosphorylation. Many, but not all, of the light-harvesting complexes are integral membrane proteins in which chlorophylls are coordinated to amino acid side chains or sometimes to water molecules, main chain carbonyl groups or lipid, within the TM helical segments of the LH complexes. The carotenoids make more non-polar interactions sometimes with a single hydrogen bond at each end, whereas iron–sulphur complexes are coordinated by cysteine side chains. In many cyanobacteria and algae, there are additional soluble accessory light-harvesting protein complexes, which bind to the membrane and further enhance the absorption of incident light photons. The direct energy transfer mechanism, which also depends on spectral overlap and transition dipole orientation, is most efficient over small distances. The time it takes is less than 1–2 ps when the chromophores are closer than 15 Å but the excited state lifetimes increase to 30–50 ps as the distances increase to 25 Å. The overall structures of the complexes have clearly evolved to provide a stable environment that optimises the energy transfer efficiency while decreasing the likelihood of adverse oxidation events.
In 1985, the purple bacterial Rhodopseudomonas viridis reaction centre structure was the first high resolution membrane protein structure to be determined (Deisenhofer et al. Reference Deisenhofer, Epp, Miki, Huber and Michel1985), and at that time was also the largest asymmetric structure ever determined by X-ray crystallography. It has 11 TM helices altogether (Fig. 19a). There are five TM helices in the L and M subunits, which are curled around each other related by a local pseudo-twofold axis perpendicular to the membrane, and one in subunit H, which is offset to one side. Subunits L and M surround the chromophores in the centre of the structure, including four chlorophyll-b molecules, two bacteriopheophytins, two quinones and an iron centre. The structure was subsequently refined (Deisenhofer et al. Reference Deisenhofer, Epp, Sinning and Michel1995) and is shown in Fig. 19a. It represents the very heart of photosynthesis since the central ‘special pair’ of bacteriochlorophyll-b molecules is the site at which the electronic excitation that follows light absorption is first converted into electronic charge separation across the width of the membrane. Similar reaction centres are found in all photosynthetic organisms, built around subunits homologous to the L and M subunits of purple bacteria. These range from the purple and green sulphur bacteria, which have only one reaction centre and do not evolve oxygen, to more complex organisms such as cyanobacteria and green plants, which have two photosystems, PSI and PSII, and generate oxygen from water (Ben-Shem et al. Reference Ben-Shem, Frolow and Nelson2003; Loll et al. Reference Loll, Kern, Saenger, Zouni and Biesiadka2005; Amunts et al. Reference Amunts, Drory and Nelson2007). In PSII, subunits D1 and D2 are homologous to L and M, whereas in PSI, the five C-terminal TM helices in PsaB and PsaA occupy the equivalent positions. In addition, the light-harvesting subunits CP43 and CP47 in PSII, each with six TM helices, are adjacent to D1 and D2, and are related by the same pseudo-twofold axis. CP43 and CP47 in turn occupy homologous positions to the six N-terminal helices of PsaB and PsaA. Beyond these central core subunits of all the reaction centres, the structures then diverge. Fig. 19b to d shows how these different but related structures have developed during evolution. At greater physical distances from the RC, the nature of the light-harvesting subunits in different organisms diverges with a clear difference then emerging between those in bacterial photosynthetic complexes and those in algae and vascular plants.

Fig. 19. Photosynthetic complexes: (a) reaction centre (1PRC), (b) RC-LH1 complex (1PYH), (c) PSI complex (2O01) and (d) PSII complex (3BZ1). The common reaction centre (RC) core can be seen here in the same orientation in each part of the figure. In (b), (c) and (d), the RCs are surrounded by a great variety of subunits (see text). The oxygen-evolving centre (OEC) is shown as pink spheres in PSII. Choloropyhll in all proteins are shown as grey sticks.
The major plant LHC, known as LHCII is arguably the most abundant membrane protein on planet Earth. It has 3 TM helices, 14 chlorophylls and 4 carotenoids, and forms trimers (Fig. 28 a). In contrast, the purple bacteria have quite different light-harvesting complexes made up of two single TM protein subunits (α and β), each of which binds three chlorophylls and two carotenoids. Bacterial LH1 forms a ring of 15 LH subunits around each reaction centre (Roszak et al. Reference Roszak, Howard, Southall, Gardiner, Law, Isaacs and Cogdell2003), whereas bacterial LH2 forms similar rings of 8 or 9 subunits that are not in direct contact with the RC. In PSI from cyanobacteria (Jordan et al. Reference Jordan, Fromme, Witt, Klukas, Saenger and Krauss2001), there is an arc of four LHCI molecules, homologous to LHCII, each with three TM helices, on the periphery of the core. Thus, two quite different structures are used for light-harvesting in the purple bacteria and in higher plants, but both funnel the light energy down an energy gradient, ending up in both cases at the RC, where the energy is used for charge separation and converted into a potential gradient across the membrane.
In one of the simplest cases, such as RC-LH1 from Rhodopseudomonas palustris, there are three RC proteins and two LH subunits, together with 50–60 chromophores. By contrast in pea or cyanobacteria, there are 17 to 20 protein subunits and 50–170 chlorophylls in addition to numerous carotenoids, iron–sulphur clusters and quinones. These structures are among the most complex known in biology to date, but show a remarkably simple overall plan, much simpler for example than the structures of the ribosome or the RNA polymerases that must perform a complex series of reactions in which the reactants are themselves macromolecules that undergo very large movements during their respective reactions. The molecular complexes of photosynthesis, in contrast, have a relatively simple task that does not require them to move. Their main role is to maintain light-absorbing and photoprotecting chromophores at exactly the right distance for efficient energy transfer and charge separation.
PSII also includes the oxygen-evolving centre with its manganese cofactor that is coordinated to residues in D1 and CP43 (see Fig. 19d) but the resolution of current structures will need to be improved to shed further light on how oxygen is evolved (Ferreira et al. Reference Ferreira, Iverson, Maghlaoui, Barber and Iwata2004; Loll et al. Reference Loll, Kern, Saenger, Zouni and Biesiadka2005).
G-protein-coupled receptors
Members of the G-protein-coupled receptor (GPCR) family are found in organisms from yeast to man. They are involved in sensory perception (smell, sight, taste) and trans-membrane signalling in hundreds of processes throughout human physiology. Recent atomic structures of the adenosine A2a receptor (A2aR) (Jaakola et al. Reference Jaakola, Griffith, Hanson, Cherezov, Chien, Lane, Ijzerman and Stevens2008) and of β1- and β2-adrenergic receptors (βAR) (Cherezov et al. Reference Cherezov, Rosenbaum, Hanson, Rasmussen, Thian, Kobilka, Choi, Kuhn, Weis, Kobilka and Stevens2007; Warne et al. Reference Warne, Serrano-Vega, Baker, Moukhametzianov, Edwards, Henderson, Leslie, Tate and Schertler2008) have joined bovine (Palczewski et al. Reference Palczewski, Kumasaka, Hori, Behnke, Motoshima, Fox, Le Trong, Teller, Okada, Stenkamp, Yamamoto and Miyano2000; Li et al. Reference Li, Edwards, Burghammer, Villa and Schertler2004) and squid rhodopsin (Murakami & Kouyama, Reference Murakami and Kouyama2008) structures to give us a good idea of the range of structural variation in the family. All show the well-known seven-helix bundle structure with strongly kinked helices 2, 4, 5, 6 and 7, frequently at the position of a conserved proline residue in the structure (Fig. 20).

Fig. 20. G-protein-coupled receptors: (a) bovine rhodopsin (1GZM), (b) β1-adrenergic receptor (2VT4), (c) β2-adrenergic receptor (2RH1) and (d) adenosine A2a receptor (3EML). These four structures all represent inactive, antagonist-bound, conformations of four different G-protein-coupled receptors. The structures are related as they all have a homologous seven-helix TM bundle, but differ in the details of their binding sites and structures at both cytoplasmic and extracellular surfaces. A hypothetical mechanism of activation and signalling to the cytoplasmic αβγ G-protein complex is described in the text.
However, only for bovine rhodopsin is direct experimental information available about the nature of the structural change that triggers G-protein activation. From double electron-electron resonance (DEER) spectroscopy (Altenbach et al. Reference Altenbach, Kusnetzow, Ernst, Hofmann and Hubbell2008) together with suggestive crystal structures of two unliganded opsins (Park et al. Reference Park, Scheerer, Hofmann, Choe and Ernst2008; Scheerer et al. Reference Scheerer, Park, Hildebrand, Kim, Krauss, Choe, Hofmann and Ernst2008), it is likely that a ~6-Å outward movement of helix 6 and smaller movements of helices 1, 5 and 7 trigger binding of the G-protein complex and subsequent signalling. The structure of opsin with a bound 11 residue C-terminal peptide from Gα (Scheerer et al. Reference Scheerer, Park, Hildebrand, Kim, Krauss, Choe, Hofmann and Ernst2008) gives a first glimpse of how the GPCR structural change might allow G-protein binding, which leads to GDP/GTP exchange and subsequent signalling. The peptide binds in a cavity that is opened up by the outward movement of helix 6, and which is non-existent in the ground state rhodopsin structure where 11-cis retinal occupies the ligand-binding site. A plausible but hypothetical mechanism of coupling the binding of this C-terminal helix (α5) of Gα to the subsequent GDP/GTP exchange has been proposed, based on the apparent necessity for a ~40° tilt of α5 out of its position in the free Gαβγcomplex (Scheerer et al. Reference Scheerer, Park, Hildebrand, Kim, Krauss, Choe, Hofmann and Ernst2008).
A comparison of the structure and interactions that high affinity antagonists make in the A2aR and βAR structures with the chemical formulae of the natural agonists suggests that the ligand binding site in GPCRs will contract to fit tightly around an agonist on the extracellular side of the receptor, whereas antagonists will stabilise the unliganded form, which is predominantly inactive except in those GPCRs with high constitutive activity. In β1AR and β2AR, the hydroxyl groups of the three serine residues on helix 5 that are known to interact with the two catecholamine hydroxyl groups of adrenaline are too distant to form direct hydrogen bonds in an atomic model (Warne et al. Reference Warne, Serrano-Vega, Baker, Moukhametzianov, Edwards, Henderson, Leslie, Tate and Schertler2008) unless the proposed agonist-induced structural change occurs, although there is also the possibility that water molecules in the ligand-binding site may mediate the interaction. A similar argument can be made for interaction of the 2′- and 3′-hydroxyl groups of the adenosine agonist in the A2a receptor with the imidizole side-chain of His-278, which is the residue homologous to the retinal Schiff base residue, Lys-296, on helix 7 in bovine rhodopsin. This ligand-induced conformational change provides the energy for similar movements of helices 6 and 7 on the intracellular side as seen in rhodopsin and bacteriorhodopsin. Although seven helix membrane proteins have a rich variety of specificities and functions, they appear also to have an underlying mechanistic unity.
Membrane enzymes
Intramembrane proteases
Recent identification of intramembrane proteases has revealed a new strategy for cellular regulation where inactive transmembrane protein precursors in their membrane bound form are activated by intramembrane proteolytic cleavage. Proteolysis results in the release of cytoplasmic, luminal or extracellular domains that move to new locations where they can carry out their biological function (Weihofen & Martoglio, Reference Weihofen and Martoglio2003; Wolfe & Kopan, Reference Wolfe and Kopan2004; Freeman, Reference Freeman2008). Different intramembrane protease families, classified as serine, aspartyl or metallo-proteases have been identified in all kingdoms of life (cysteine intramembrane proteases have not yet been found). Intramembrane proteases have multiple TM helices with active site residues sequestered within distinct TM domains. Intramembrane proteolysis has so far been implicated in diverse cellular processes including differentiation, the unfolded protein response, lipid metabolism, signal peptide processing and, in prokaryotes, generation of peptide pheromones, response to extracellular stress and pathogenesis (Wolfe & Kopan, Reference Wolfe and Kopan2004; Freeman, Reference Freeman2008). Structures of intramembrane proteases are important for understanding how catalysis occurs within the membrane using the same or similar mechanisms to those used by soluble proteases.
Rhomboids
Rhomboids are six TM domain serine proteases, the first of which was identified originally as a component of the EGF receptor-signalling pathway in Drosophila and named after the appearance of wing vein patterns in developmental mutants. Since then they have been shown to play a major role in bacterial quorum sensing and protozoal pathogenesis (Urban et al. Reference Urban, Lee and Freeman2001; O'Donnell et al. Reference O'Donnell, Hackett, Howell, Treeck, Struck, Karnajski, Withers-Martinez, Gilberger and Blackman2006; Stevenson et al. Reference Stevenson, Strisovsky, Clemmer, Bhatt, Freeman and Rather2007). In vivo and in vitro cleavage assays of different rhomboids have identified key residues in catalysis and point to a serine-histidine dyad, similar to that formed in the subtilisin protease family (Urban et al. Reference Urban, Schlieper and Freeman2002; Lemberg et al. Reference Lemberg, Menendez, Misik, Garcia, Koth and Freeman2005; Urban & Wolfe, Reference Urban and Wolfe2005). Since the hydrolysis of peptide bonds requires water, the concept of proteolysis in the membrane has remained uncertain, particularly the question of whether the active site resides within the membrane or at the surface where water molecules are plentiful. The crystal structures of prokaryotic rhomboids from E. coli and Haemophilus influenzae show how this is achieved (Wang et al. Reference Wang, Zang and Ha2006; Wu et al. Reference Wu, Yan, Feng, Oberstein, Yan, Baker, Gu, Jeffrey, Urban and Shi2006; Ben-Shem et al. Reference Ben-Shem, Fass and Bibi2007; Lemieux et al. Reference Lemieux, Fischer, Cherney, Bateman and James2007). In the core, a relatively short TM helix 4 and an extended loop 3 is surrounded by other helices to create a hydrophilic cavity that opens to the periplasmic side (Fig. 21a). The active site serine is located ~10 Å into the bilayer and hydrogen bonded to the conserved histidine. Numerous water molecules are found in this cavity that is capped by flexible loop 5. It is likely that this loop can flip spontaneously (Wang & Ha, Reference Wang and Ha2007; Wang et al. Reference Wang, Maegawa, Akiyama and Ha2007). An interesting feature of the rhomboid structure is that loop 1 is placed outside the helix bundle, and forms a hairpin that is partially buried in the membrane. Mutation in this loop renders the enzyme inactive, but its exact function remains to be determined. Four structures of rhomboids have been solved in different detergents and space groups and their comparison has helped to explain the possible mechanism of substrate recruitment. The greatest variation is observed in helix 5, a non-conserved TM helix exposed to the lipid bilayer. Together with loop 5, helix 5 may be involved in recruitment of substrate and presenting it to the active site. In the absence of a rhomboid structure with a substrate or inhibitor, the mechanism of catalysis and regulation remains largely unknown.

Fig. 21. Structure of intramembrane proteases: (a) rhomboid GlpG (2IC8) and (b) site-2 protease (3B4R). (a) Rhomboids are serine proteases that utilise a catalytic dyad of serine and histidine (red) for hydrolysis of peptide bonds in the membrane. GlpG, a prokaryotic rhomboid from E. coli represents the core architecture with a bundle of six TM helices. A relatively short helix 4 and extended loop 3 (yellow) are placed at the centre of the molecule surrounded by other TM helices to form a hydrophilic cavity with numerous water molecules. Loop 5 caps this active site (orange). Loop 1, an essential component but whose function is unknown lies outside of the helical bundle (magenta). TM helices 1, 3 and 6 provide a structural support (green). Substrate is thought to enter through an opening between TM helices 2 and 5 (blue). (b) Site-2 protease from M. jannaschii is a zinc containing metalloprotease of unknown biological function. Zinc (red sphere) is coordinated by conserved histidines and aspartate (magenta). It is likely to be in a tetrahedral coordination with a water molecule (not observed due to limited resolution) forming the fourth coordination. TM helices 2, 3 and 4 (yellow) form the core that holds the key residues for catalysis. Discontinuous part of helix 4 is shown in brown. TM1 with a mixture of α-helix and β-strand is shown in blue. Outer helices TM 5 and 6 are in green.
Site-2 protease
First identified in the regulation of cholesterol biosynthesis and uptake, site-2 proteases (S2P) are also involved in ER stress signalling, response to periplasmic stress in E. coli, and sporulation in Bacillus subtilis (Brown & Goldstein, Reference Brown and Goldstein1999; Rudner et al. Reference Rudner, Fawcett and Losick1999; Ye et al. Reference Ye, Rawson, Komuro, Chen, Dave, Prywes, Brown and Goldstein2000; Yu & Kroos, Reference Yu and Kroos2000; Alba et al. Reference Alba, Leeds, Onufryk, Lu and Gross2002). In contrast to rhomboids that do not require substrate trimming, substrate proteolysis by S2P requires prior cleavage by another protease called site-1 protease. S2P contains the signature HExxH sequence. The active site zinc is coordinated by histidines on one TM helix together with a conserved aspartate in another TM helix. These active site residues resemble those in thermolysin, a soluble metalloprotease. Activation by glutamate of a water molecule bound to zinc results in nucleophilic attack on the scissile peptide bond. These conserved sequences and the zinc ion are predicted to be within the bilayer.
The crystal structure of the core domain of S2P from M. jannaschii (MjS2P) at 3·3 Å reveals a mixture α-helices and β-strands (Feng et al. Reference Feng, Yan, Wu, Yan, Wang, Jeffrey and Shi2007). As expected for a metalloprotease, zinc is found coordinated to the histidines and aspartate. The present structure with its approximate membrane boundary places them ~14 Å into the bilayer (Fig. 21b). Six TM helices form the core of MjS2P. The conserved HExxH motif is found in TM2. Helix 4 is not continuous but is divided into two halves (H4a and H4b) by a nine-amino acid bulge. The aspartate that coordinates zinc is found at the N-terminus of H4b. Such discontinuous helices are not uncommon in membrane proteins but are most frequently found in transport proteins where they transiently coordinate ions. A conserved asparagine is placed close to the active site, which might be essential to neutralise the negative charge during hydrolysis.
Two independent molecules in the asymmetric unit of Mj2P show remarkable differences in the outer helices TM1 and 6 and shed light on possible structural changes during activity. It has been speculated that these molecules might represent open and closed states. Movement of these helices, like that of TM5 in rhomboids, has been proposed to act as a gate for substrate binding and release. An intriguing feature in the structure of MjS2P is the TM1 segment that has been modelled to consist of an α-helix and a β-strand. Such a mixture of secondary structure in the lipid bilayer is very unusual for a membrane protein. β-Strands have been found in other membrane proteins but only at the interface between bilayer and bulk solution. Hence, it is hard to imagine how a mixed structure as in MjS2P can form a stable entity in the lipid bilayer. However, structures of membrane proteins have provided many surprises before, so it may be true that MjS2P does have a mixed structure in TM1. Could this be an effect of crystallisation in detergent or crystal packing? As the authors have pointed out, the flexibility seen in TM1 and TM6 might be an effect of non-native detergent environment. It is therefore essential to determine a structure in the lipid bilayer so that the exact nature of TM1 and probably TM6 can be confirmed, and the mechanism of proteolysis explored.
Thiol oxidases
Formation of disulphide bonds is important for stability and correct folding of many proteins, in particular those that are secreted. In Gram-negative bacteria, the catalysis of disulphide bond formation occurs in the periplasmic space. In E. coli DsbA, a highly reactive thiol oxidase rapidly introduces disulphide bonds by thiol exchange into newly folding proteins (Inaba & Ito, Reference Inaba and Ito2008). DsbA consists of a thioredoxin-like domain that harbours the conserved active site (C30-P-X-C33) and an α-helical insertion domain. Reoxidation of DsbA is an essential process and a membrane enzyme DsbB converts the oxidising equivalent of quinones into reactive disulphides thereby connecting oxidative protein folding to respiratory pathways. DsbB is a left-handed four TM α-helix bundle that exposes two pairs of cysteines on its two periplasmic loops (Fig. 22a). Cysteine 41 and 44 are located in periplasmic loop 1 (PL1) while cysteine 104 and 130 are located in loop 2 (PL2). DsbB and DsbA are dynamic enzymes that can form intramolecular and intermolecular disulphide bonds resulting in various intermediate states. It is thought that co-ordinated action of four cysteines in DsbB together with ubiquinone results in oxidation of DsbA. Thus, it is possible to trap intermediates by mutating cysteine residues to form a stable DsbB-DsbA complex or a reaction intermediate with an interloop disulphide bond between C41 and C130 of DsbB. Multiple structures of DsbB alone or in complex with DsbA have been determined by X-ray crystallography and NMR spectroscopy providing complementary structural information (Inaba et al. Reference Inaba, Murakami, Suzuki, Nakagawa, Yamashita, Okada and Ito2006; Malojcic et al. Reference Malojcic, Owen, Grimshaw and Glockshuber2008; Zhou et al. Reference Zhou, Cierpicki, Jimenez, Lukasik, Ellena, Cafiso, Kadokura, Beckwith and Bushweller2008; Inaba et al. Reference Inaba, Murakami, Nakagawa, Iida, Kinjo, Ito and Suzuki2009). The NMR structure shows an N-terminal amphipathic helix that lies parallel to the membrane, which is disordered in the crystal structure. Ubiquinone binds in a groove formed between TM1 and TM4 close to the cysteines in PL1 (Fig. 22a). The most important region in DsbB is the periplasmic loop that can be divided into distinct regions starting with a N-terminal region that lacks any secondary structure and has high mobility. This region is followed by an amphiphatic helix that is thought to reside in the bilayer. This helix is then followed by PL2 that lies on top of TM2 so that interloop disulphides between C41 and C130 can be formed (Zhou et al. Reference Zhou, Cierpicki, Jimenez, Lukasik, Ellena, Cafiso, Kadokura, Beckwith and Bushweller2008).
Fig. 22. Membrane enzymes (a) thiol oxidase DsbB-DSbA (2ZUP), (b) leukotriene LTC4 synthase trimer (2UUH) and (c) methane monooxygenase MMO (1YEW). Some enzymes are integral membrane proteins because they have hydrophobic substrates or because they are coupled to components of the electron transport chain. DsbB-DsbA complex: Disulphide bond formation in nascent polypeptides is catalyzed in the periplasm of Gram-negative bacteria by the highly reactive enzyme DsbA. DsbB is a left-handed four TM α-helical membrane protein that regenerates DsbA by reduction of a disulphide bond, followed by subsequent thiol oxidation in DsbB by ubiquinones. Conserved cysteine residues in DsbA (cys 30 and 33) and DsbB (cys 41, 44, 104, 130) play a crucial role in regeneration of DsbA by thiol exchange mechanism. It is possible to trap reaction intermediates of the DsbB-DsbA complex by mutating specific cysteine residues. The figure depicts one such intermediate where Cys33 is mutated to alanine in DsbA and Cys130 to serine in DsbB (both residues shown as green sticks). This results in an intramolecular disulphide bond (red) between Cys30 of DsbA and Cys104 of DsbB. Lack of cysteine at position 130 results in inter molecular disulphide between Cys41 and Cys44 of DsbB (orange). Ubiquinone (brown) is found in a groove between TM1 and TM4 close to the Cys41 and Cys44. When Cys33 of DsbA attacks the disulphide bond between DsbB-DsbA, it releases oxidised DsbA. Subsequent thiol exchange within DsbB restores its oxidised state. LTC4 synthase: Membrane proteins involved in eicosanoid and glutathione metabolism are collectively called MAPEG (membrane associated proteins in eicosianid and glutathione metabolism). They play a key role in the generation of lipid mediators from arachidonic acid, which is responsible for bronchoconstriction leading to asthma. In addition, they play a major role in oxidative stress and xenobiotic detoxification. The membrane enzyme LTC4 synthase converts leukotriene A4 to leukotriene C4 by covalent coupling to glutathione. Like other MAPEG family members, LTC4 synthase is trimeric, with each monomer (cyan, blue and green) having four TM helices. Glutathione (red) binds at a crevice between two monomers. A detergent molecule (orange) that might mimic a substrate is shown close to glutathione. The structure of LTC4 synthase suggests how glutathione is activated and coupled to the substrate but a definitive answers await more biochemical and probably more structural data. Methane monooxygenase: due to its inert nature, industrial oxidation of methane is an extremely difficult process. Methanotropic bacteria that use methane as their sole carbon source have evolved enzymes to carry out this difficult oxidation process. Membrane bound particulate methane monooxygenase is a homotrimer of three subunits giving a molecule with nine chains altogether (one monomer is shown in colour). Subunits A (dark blue) and C (yellow) together make up most of the TM domain with 12 (7 from subunit A and 5 from subunit C) helices. Subunit B (magenta) has two TM helices and a large soluble domain that contains two β-barrels. Three metal centres are observed in the crystal structure. A mononuclear copper (red) and a dinuclear copper centre (brown) are found in the soluble barrel domain of subunit B. A zinc ion (green) is found in the membrane coordinated by residues from subunit A and C. These metal centres must play a major role in activation of oxygen and subsequent generation of methanol.
Interaction of DsbA with DsbB induces a conformational change in PL2 while very little change in observed in the rest of protein. The N-terminal section of PL2 that harbours C104 is accommodated close to C30 of DsbA. This interaction results in separation of DsbB C130 from C104 and positions it close to C41 for a nucleophilic attack and subsequent disulphide bond formation. When C33 of DsbA attacks the DsbB-C104 and DsbA-C30 disulphide, oxidised DsbA is released while subsequent disulphide rearrangement in four cysteines of DsbB aided by ubiquinone restores DsbB for yet another cycle.
Membrane associated proteins in eicosanoid and glutathione metabolism
Lipid mediators derived from the arachidonic acid (AA) cascade are crucial in the defence repertoire of immune cells. Molecules derived from AA mediate processes of inflammation, fever and pain. Activation of phospholipase A2 results in release of arachidonic acid, which can be converted by cyclooxygenase, a membrane anchored enzyme, into prostanoids such as prostaglandins or thromboxane. Alternatively, AA can be converted into 20-carbon unsaturated fatty acid compounds called leukotrienes (LT) (Bisgaard, Reference Bisgaard2001). Enzymes involved in the conversion of AA to LT are called Membrane Associated Proteins in Eicosanoid and Glutathione metabolism (MAPEG). In humans, the MAPEG family includes the leukotriene C4 synthase (LTC4S), microsomal prostaglandin E synthase 1 (mPGES1), 5-lipooxygenase activating protein (FLAP) and the microsomal glutathione S-transferase (mGST). FLAP has no enzyme activity but provides AA to 5-lipooxygenase. LTC4S is responsible for generation of cysteinyl-leukotriene, a potent broncho-constricting agent that is upregulated in asthma (Zhu et al. Reference Zhu, Qiu, Figueroa, Bandi, Galczenski, Hamada, Guntupalli, Evans and Jeffery2005). There are other cysteinyl-leukotrienes downstream of LTC4 that are not made directly by LTC4S. mGST enzymes respond to oxidative stress and detoxify xenobiotics (Martinez Molina et al. Reference Martinez-Molina, Eshaghi and Nordlund2008).
Proteins belonging to the MAPEG family are trimeric, each monomer having four TM helices. Except for FLAP, which does not show a glutathione transferase activity, other members possess an Rx3Nx2E/D motif involved in binding of glutathione. Electron crystallographic studies of mGST1 provided the first structural information on the architecture of the MAPEG family (Schmidt-Krey et al. Reference Schmidt-Krey, Mitsuoka, Hirai, Murata, Ceng, Fujiyoshi, Morgenstern and Hebert2000; Holm et al. Reference Holm, Bhakat, Jegerschold, Gyobu, Mitsuoka, Fujiyoshi, Morgenstern and Hebert2006), followed by the X-ray structures of LTC4 synthase (Ago et al. Reference Ago, Kanaoka, Irikura, Lam, Shimamura, Aausten and Miyano2007; Martinez Molina et al. Reference Martinez-Molina, Wetterholm, Kohl, Mccarthy, Niegowski, Ohlson, Hammarberg, Eshaghi, Haeggstrom and Nordlund2007), FLAP (Ferguson et al. Reference Ferguson, Mckeever, Xu, Wisniewski, Miller, Yamin, Spencer, Chu, Ujjainwalla, Cunnighham, Evans and Becker2007) and the recent EM structure of mPGES1 (Jegerschold et al. Reference Jegerschold, Pawelzik, Purhonen, Bhakat, Gheorghe, Gyobu, Mitsuoka, Morgenstern, Jakobsson and Hebert2008). The overall TM helical fold is similar in all three proteins. The structure of FLAP with bound inhibitor deviates considerably from other structures in the family at the C-terminus of TM4, which could be induced by inhibitor binding. LTC4 synthase has an additional amphiphatic helix that lies outside the putative membrane boundary and may interact with lipid headgroups.
Glutathione (GSH) is buried between adjacent monomers, with its γ-carboxylate group facing the polar residues of protein and its thiol group facing the bilayer. This site is close to the cytoplasmic face of the outer nuclear membrane and ideally placed for release of products (Fig. 22b). It is covered by the loop connecting helices TM1 and TM2, whose flexibility might be required during the catalytic cycle. There has been speculation that a detergent molecule found close to the glutathione-binding site in the structure of LTC4S might resemble a substrate molecule. Conserved arginines may play a role in enhancing the nucleophilicity of GSH and its subsequent conjugation to the substrate molecule. The TM helices of MAPEG show similarities to subunit I of cytochrome oxidase indicating a common evolution of proteins involved in oxidative stress and electron transfer.
Methane monooxygenase
Many enzymes from prokaryotes and particularly archaea carry out very difficult chemical reactions, which have the potential to be used in industrial applications and bioremediation. One such enzyme is methane monooxygenase that catalyzes the oxidation of methane (Sullivan et al. Reference Sullivan, Dickinson and Chase1998; Hakemian & Rosenzweig, Reference Hakemian and Rosenzweig2007). Natural gas (principally methane) as an alternative fuel has gained a lot of attention but the industrial process of methane oxidation is extremely difficult. Methanotropic bacteria use methane as their sole carbon source and oxidise it to methanol in the first step of metabolism. Methane monooxygenase (MMO) is found either as a soluble enzyme or as membrane bound particulate methane monooxygenase (pMMO). The levels of the soluble and the membrane bound forms are regulated by level of copper in the environment (Hakemian & Rosenzweig, Reference Hakemian and Rosenzweig2007). Understanding pMMO at the molecular level may help design catalysts for the oxidation of methane.
The structure of pMMO reveals a homotrimer of three different subunits A, B and C, making nine polypeptides in each molecule (Lieberman & Rosenzweig, Reference Lieberman and Rosenzweig2005). Subunit B forms much of the soluble domain with a seven-stranded N-terminal β-barrel and an eight-stranded C-terminal β-barrel. The barrels are connected to two TM helices by a long loop. Subunits A and C with seven and five TM helices, respectively, comprise much of the TM domain. The TM helices of subunit A pack against the TM helices of subunit B. Three metal centres are observed in the crystal structure. A mononuclear copper centre is found at the surface of the N-terminal barrel co-ordinated by histidines, 25 Å from the membrane. Also in the N-terminal barrel is a dinuclear copper centre with similarities to cytochrome oxidase CuA, ~10 Å from the membrane. A third metal centre in the membrane is a zinc ion, co-ordinated by residues from subunits A and C, and lies ~19 Å away from the dinuclear copper centre (Fig. 22c). The observation of zinc in the structure may be due to the high concentration used in crystallisation. In vivo, this is probably occupied by copper or iron. It is not clear from the structure how oxidation of methane is catalyzed (Balasubramanian & Rosenzweig, Reference Balasubramanian and Rosenzweig2007). Obviously one of these metal centres is involved in activation of oxygen but at this stage there is not enough experimental data to identify which metal centre is the active site.
Monotopic membrane proteins
Although this review is focussed on integral, membrane-spanning proteins, there is a considerable number of membrane proteins that interact strongly with membranes, while penetrating only slightly into the surface of the lipid bilayer. The best definition of such monotopic membrane proteins is that they can be released from the membrane only using detergent, yet do not span the full width of the lipid bilayer. These proteins can be anchored to the membrane in different ways. For example, they can interact with the lipid headgroups via an amphipathic helix that is oriented parallel to the surface of the bilayer, or they can have a covalently attached lipid chain, such as a palmitoyl group or glycosylphosphatylinositol (GPI) or they can have multiple ionic interactions. The enzyme prostaglandin synthetase (Picot et al. Reference Picot, Loll and Garavito1994) is monotopic and interacts with the bilayer through a hydrophobic patch on the surface of the protein. Similarly cytochrome P450 enzymes (Yano et al. Reference Yano, Wester, Schoch, Griffin, Stout and Johnson2004) are found in microsomal or mitochondrial inner membranes and normally have an N-terminal, non-cleavable membrane insertion sequence, which acts as an anchor. Carnitine palmitoyltransferase is another monotopic membrane protein (Rufer et al. Reference Rufer, Thoma, Benz, Stihle, Gsell, De Roo, Banner, Mueller, Chomienne and Hennig2006), which interacts via hydrophobic and positively charged residues (Rufer et al. Reference Rufer, Lomize, Benz, Chomienne, Thoma and Hennig2007). Glycerol-3-phosphate dehydrogenase, a flavin-linked enzyme in E. coli, essential for aerobic growth in glycerol (Yeh et al. Reference Yeh, Chinte and Du2008), is another example. Some proteins whose active sites are outside the membrane have a predicted TM helix that probably spans the membrane but is not essential for activity. Structures of some of these proteins with the TM helix deleted include monoamine oxidase (Binda et al. Reference Binda, Newton-Vinson, Hubalek, Edmondson and Mattevi2002), signal peptide peptidase (Kim et al. Reference Kim, Oliver and Paetzel2008) and peptidoglycan glycosyltransferase (Lovering et al. Reference Lovering, De Castro, Lim and Strynadka2007).
β-Barrel membrane proteins
Although α-helical structures dominate in membrane proteins (Fig. 1) there exist membrane proteins with β-strands that, due to restrictions posed by the hydrophobic lipid bilayer, invariably form barrels. The primary sequences of most β-barrels show non-polar amino acids at every second residue, facing the hydrophobic lipid environment, while the interior of the barrels are generally polar creating hydrophilic channels. However, in some cases, the interior is non-polar in proteins that have evolved to transport hydrophobic substrates. Thus, the environment in the interior of the barrel allows transport through the centres of the barrels and therefore through the membrane. This arrangement is in contrast to the β-barrels of soluble proteins, which bury their non-polar residues at the core of their structure while exposing polar residues to the aqueous exterior. β-Barrel membrane proteins dominate the outer membrane of Gram-negative bacteria. They carry out a wide range of functions that include the transport of macromolecules such as protein, DNA, polysaccharides and lipids. They also act as receptors for bacteriophages. A few are membrane-anchored enzymes and some are involved in extrusion of drugs and antibiotics (Nikaido, Reference Nikaido2003). Wza, a polysaccharide transporter is the only known α-helical membrane spanning protein in the outer membrane of Gram-negative bacteria (Dong et al. Reference Dong, Beis, Nesper, Brunkan-Lamontagne, Clarke, Whitfield and Naismith2006). A possible explanation for the large number of β-barrels observed in the outer membrane has been attributed to their less hydrophobic nature, which might aid in efficient transport across inner membrane and periplasmic space without the risk of aggregation. Crystallisation of β-barrel outer membrane proteins has been found to be much easier than α-helical TM proteins, largely due to their inherent stability. Numerous structures of β-barrels have been solved even with protein refolded from inclusion bodies (Gouaux, Reference Gouaux1998; Buchanan, Reference Buchanan1999; Schulz, Reference Schulz2002). The smallest β-barrel membrane protein structure known so far has 8 strands (the smallest β-barrel found in soluble proteins has 6 strands) while the largest has 24 (Remaut et al. Reference Remaut, Tang, Henderson, Pinkner, Wang, Hultgren, Thanassi, Waksman and Li2008).
Since the outer membrane of Gram-negative bacteria has no proton motive force, most proteins act as passive pores. Some of them form non-selective channels such as OmpF and OmpC porins with 16-stranded barrels. They transport molecules up to 600 Daltons and show a slight preference for cations (Cowan et al. Reference Cowan, Schirmer, Rummel, Steiert, Ghosh, Pauptit, Jansonius and Rosenbusch1992; Basle et al. Reference Basle, Rummel, Storici, Rosenbusch and Schirmer2006). It has also been shown that OmpF can transport unfolded polypeptides, a property utilised by colicins (Yamashita et al. Reference Yamashita, Zhalnina, Zakharov, Sharma and Cramer2008). Porins such as LamB (Fig. 23) and ScrY have 18-stranded β-barrels yet have more restricted pores and transport carbohydrates (Schirmer et al. Reference Schirmer, Keller, Wang and Rosenbusch1995; Forst et al. Reference Forst, Welte, Wacker and Diederichs1998). These classical porins form SDS stable homotrimers, each monomer having a separate translocation pathway. Maltoporins have a string of low affinity sugar binding sites lined with aromatic residues termed the greasy slide, which allows the rapid uptake oligosaccharides. Not all outer membrane proteins form pores. Some have evolved to perform other biological functions while closing the interior of the barrel with an intricate hydrogen-bonding network. Proteins with smaller barrels such as OmpX (Vogt & Schulz, Reference Vogt and Schulz1999) act as membrane anchors or for adhesion while OmpA, an abundant protein in E. coli, stabilises the outer membrane and has been used extensively to study protein folding (Pautsch & Schulz, Reference Pautsch and Schulz1998). There also exist many outer-membrane proteins with no known biological functions, one of which is the 14-stranded β-barrel OmpG, regulated by pH (Yildiz et al. Reference Yildiz, Vinothkumar, Goswami and Kuhlbrandt2006). Using the β-barrel as an anchor in the membrane, proteins have also evolved to carry out enzymatic reactions. OmpT, a protease whose catalytic centre protrudes from a 10-stranded β-barrel to the external medium, is involved in protein degradation (Vandeputte-Rutten et al. Reference Vandeputte-Rutten, Kramer, Kroon, Dekker, Egmond and Gros2001) and OmpLA (Fig. 24a), a phospholipase that cleaves lipopolysaccharides, forms a 12-stranded barrel (Snijder et al. Reference Snijder, Ubarretxena-Belandia, Blaauw, Kalk, Verheij, Egmond, Dekker and Dijkstra1999).

Fig. 23. Architecture of porins (a) LamB trimer top view and (b) LamB monomer side view (1MAL). Found abundantly in the outer membrane of Gram-negative bacteria, β-barrel membrane proteins form the second major structural class. Depicted in the figure is a representative outer membrane porin LamB, a trimeric sugar transporter. Each monomer has an individual transport pathway and transports maltodextrin (red space filling model). LamB is an 18-stranded β-barrel with a pore that selects carbohydrates through interaction with aromatic amino acids. Other classical porins such as OmpF, OmpC, PhoE are also trimers but have 16 β-strands. All these proteins act as passive pores. The trimers of these proteins show remarkable stability even in presence of SDS.

Fig. 24. β-Barrel membrane proteins with diverse biological function (a) OmpLA (1QD5), (b) FepA (1FEP), (c) NalP (1UYN) and (d) VDAC (3EMN). β-Barrel membrane proteins are not just pore formers. They have evolved to act as receptors, enzymes and to transport specific molecules. OmpLA (a), is 12 stranded β-barrel that act as a phospholipase. OmpLA is active only as a dimer, this figure shows a monomer with its active site serine (red sticks) in complex with an inhibitor hexadecane sulphonyl fluoride (blue sticks). Transport of molecules such as iron and vitamin B12 against a concentration gradient requires energy. Since the outer membrane is devoid of any proton motive force, energy is derived from the inner membrane by transient protein-protein interactions, called the TonB-dependent transport system. Outer membrane proteins that transport these substrates are typically 22-stranded β-barrels. FepA, an iron transporter, is an example (b) of a TonB-dependent transporter. The N-terminal TonB binding domain (purple) of this protein acts as a plug to prevent leakage. Substrate binding to the outer membrane protein results in a conformational change of the TonB binding domain that triggers binding of TonB and subsequent release of substrate. Encoded in a single polypeptide chain, autotransporters have a pore forming β-barrel and a passenger domain that is released to external medium by self-cleavage. NalP from Neisseria meningitis is one such protein (c) that shows the N-terminal passenger domain (red). β-barrel proteins are also found in the outer membranes of mitochondria and chloroplast. The structure of the voltage dependent anion channel (VDAC) from mitochondria reveals a 19-stranded β-barrel. Although, adjacent β-strands in trans-membrane proteins normally pair in antiparallel orientation, the first and last strands of VDAC are parallel, which is the first time this has been observed in a β-barrel membrane protein. An N-terminal helix (yellow) is found submerged halfway through the barrel. This helix has been implicated in voltage sensing, but the biological role of membrane potential in mitochondrial outer membranes and the orientation of VDAC is unclear.
Despite lack of an energy source, some outer membrane proteins can perform active transport by using the energy derived from the cytoplasmic membrane through formation of a transient complex with inner membrane proteins. These are collectively referred to as belonging to the TonB-dependent transport system. Such proteins are comprised of an outer membrane receptor (typically a 22-stranded β-barrel) and inner membrane proteins TonB and ExbBD. Substrates such as vitamin B12 or iron are scarce in the environment and need to be scavenged by bacteria. Bacteria synthesise and secrete siderophores that have high affinity for iron and can extract Fe3+ from proteins. Vitamin B12 or iron-siderophore complexes bind to the outer membrane protein, which has an N-terminal domain (also called TonB binding domain) that acts as a plug or cork to block the opening on the periplasmic side (Ferguson et al. Reference Ferguson, Hofmann, Coulton, Diederichs and Welte1998; Locher et al. Reference Locher, Rees, Koebnik, Mitschler, Moulinier, Rosenbusch and Moras1998; Buchanan et al. Reference Buchanan, Smith, Venkatramani, Xia, Esser, Palnitkar, Chakraborty, Van der Helm and Deisenhofer1999; Ferguson et al. Reference Ferguson, Chakraborty, Smith, Esser, Van der Helm and Deisenhofer2002; Chimento et al. Reference Chimento, Mohanty, Kadner and Wiener2003; Krieg et al. Reference Krieg, Huche, Diederichs, Izadi-Pruneyre, Lecroisey, Wandersman, Delepelaire and Welte2009). The stretch of sequence in the Ton-B binding domain that binds TonB protein is called the ‘Ton-B box’. When substrates such as vitamin B12 or iron complexes bind, a conformational change in the N-terminal domain allows the binding of TonB protein from the periplasm that is accompanied by release of substrate as in FepA (Fig. 24b). Specific periplasmic proteins then bind and deliver the substrate to dedicated inner membrane transporters for its passage to the cytoplasm. The exact mechanism of energy transfer to the outer membrane β-barrel from the inner membrane remains to be elucidated.
The presence of an outer membrane provides the bacterium with a definitive barrier against foreign particles but it also means that the secretion of macromolecules requires sophisticated machinery to cross two membranes. At least six different transport systems, designated as Type I to VI secretion systems, have been reported in Gram-negative bacteria. These range from multi-subunit complexes to simplistic single protein components (Chen et al. Reference Chen, Christie and Dubnau2005; Bingle et al. Reference Bingle, Bailey and Pallen2008; Gatsos et al. Reference Gatsos, Perry, Anwari, Dolezal, Wolynec, Likic, Purcell, Buchanan and Lithgow2008). In type III and IV secretion systems, the architecture extends beyond the outer membrane towards the target cells to release effector molecules (Chen et al. Reference Chen, Christie and Dubnau2005; Marlovits et al. Reference Marlovits, Kubori, Lara-Tejero, Thomas, Unger and Galan2006). The type I and V systems have relatively simple architecture with three and one subunit protein components, respectively. The type I system has an inner membrane transporter, a fusion protein and outer membrane β-barrel. TolC, a multifunctional membrane protein involved in extrusion of drugs and proteins, forms the outer membrane component of the type I system in E. coli. Each monomer of TolC contributes four strands to form a 12-stranded barrel (Koronakis et al. Reference Koronakis, Sharff, Koronakis, Luisi and Hughes2000). The interior of the barrel is wide and without any constriction. The periplasmic domain of TolC forms a α-helical tunnel with an internal cavity of ~20 Å diameter. This domain interacts with inner membrane transport proteins such as AcrB (mentioned in section 3.2) or HlyB, either directly or through a periplasmic protein, allowing the bacterium to translocate appropriate polypeptides through the periplasm.
The type V transport system involves a single polypeptide chain that forms a β-barrel translocation domain and a passenger domain that is transported. After the passenger domain has crossed the outer membrane, self-cleavage results in its release to the external medium as in NalP (Fig. 24c). Multiple structures of translocation domains (β-barrels with 12 strands) have been reported with a α-helix from the passenger domain residing in the barrel (Oomen et al. Reference Oomen, Van Ulsen, Van Gelder, Feijen, Tommassen and Gros2004; Meng et al. Reference Meng, Surana, St Geme and Waksman2006; Barnard et al. Reference Barnard, Dautin, Lukacik, Bernstein and Buchanan2007). It is unclear how the transport occurs, but the β-barrel after the translocation of the passenger domain remains in the outer membrane with no apparent function. Since they do not require a signal sequence and probably no other protein component for transport, they have been called autotransporters.
On a similar theme, there also exist two-partner secretion pathways where the β-barrel and the passenger domain are secreted as separate entities rather than as a single polypeptide observed in autotransporters. This superfamily includes TpsB transporters where TpsB forms the β-barrel and is involved in secretion of large ‘TpsA’ proteins involved in various biological function including adhesion, cytolysis and host cell invasion as well as proteins involved in folding and insertion of outer-membrane proteins (Omp85 family). Presence of polypeptide-transport-associated (POTRA) domains at their N-terminus is a unique feature that is postulated to aid in protein interaction. FhaC, a member of Omp85-TpsB superfamily, has a 16-stranded β-barrel with a 20 residue long α-helix traversing the barrel (Clantin et al. Reference Clantin, Delattre, Rucktooa, Saint, Meli, Locht, Jacob-Dubuisson and Villeret2007). It is thought that the interaction of TpsA protein through POTRA domain promotes the translocation and the α-helix acts as a plug subsequent to translocation.
β-barrel membrane proteins have also been found in membranes of mycobacteria and related bacteria. The structure of MspA from Mycobacterium smegmatis reveals an octamer forming a 16-stranded β-barrel (Faller et al. Reference Faller, Niederweis and Schulz2004). The interior of the barrel is polar and probably creates a path for mycobacterial drug efflux. A recent addition to the growing list of β-barrel proteins is an outer membrane protein from the thermophilic bacterium Thermus thermophilus, called TtoA (Brosig et al. Reference Brosig, Nesper, Boos, Welte and Diederichs2009). It is an eight-stranded β-barrel predicted to function in adhesion.
Endo-symbiotic theory predicts the existence of β-barrels in the outer membranes of mitochondria and chloroplast. The first structure of a eukaryotic β-barrel, the voltage-dependent anion channel (VDAC) from mitochondria, proved their existence (see Fig. 24d). VDAC has been implicated in mitochondrial permeability with a role in prevention of apoptosis. Structure of VDAC determined by X-ray crystallography and NMR spectroscopy reveals a 19-stranded β-barrel, a unique feature since most β-barrel membrane proteins known thus far have an even number of strands (Bayrhuber et al. Reference Bayrhuber, Meins, Habeck, Becker, Giller, Villinger, Vonrhein, Griesinger, Zweckstetter and Zeth2008; Hiller et al. Reference Hiller, Garces, Malia, Orekhov, Colombini and Wagner2008; Ujwal et al. Reference Ujwal, Cascio, Colletier, Faham, Zhang, Toro, Ping and Abramson2008). The first and last strands of VDAC are paired parallel to each other (strands are antiparallel in all other β-barrels). The N-terminal segment of VDAC adopts a helical conformation submerged halfway through the pore and restricts the cavity (Fig. 24d). The N-terminal segment has been implicated in voltage sensing. A possible interaction site on VDAC for Bcl-XL, an anti-apoptotic protein has been revealed by NMR spectroscopy (Hiller et al. Reference Hiller, Garces, Malia, Orekhov, Colombini and Wagner2008).
Magnesium transport proteins
Magnesium is an essential cation involved in various biochemical reactions. It is the main counter ion for GTP and ATP and it maintains the structural integrity of membranes and ribosomes. The majority of Mg2+ in the cytoplasm exists in complex with nucleotides, but the free ionic concentration is ~0·5 mm. In solution, Mg2+ forms hexacoordinated bonds with the oxygen of water molecules and has the largest hydrated radius (4·76 Å) of any biologically relevant cation, but the smallest ionic radius (0·65 Å). In comparison to other cations such as Na+, K+ or Ca2+, the rate of water exchange in hydrated Mg2+ is very slow (Maguire & Cowan, Reference Maguire and Cowan2002). Such a rigid structure of Mg2+ is utilised in many biological reactions but a protein that transports Mg2+ requires a mechanism to strip strongly bound water molecules and transport a small-dehydrated cation. Mg2+ transport proteins have been classified into three families based on sequence homology, namely CorA, MgtE and MgtA/B. Of these only the MgtA/B family has an ATP binding domain and members have been classified as P-type ATPases. CorA and MgtE are probably channels but this remains to be verified (Kehres & Maguire, Reference Kehres and Maguire2002; Maguire, Reference Maguire2006).
CorA
The superfamily of CorA includes proteins of prokaryotic origin (identified as cobalt resistant mutants), mitochondrial Mrs2 and plasma membrane Alr1p (for aluminium resistance). The CorA superfamily is characterised by the presence of GMN and MPEL motifs. The crystal structure of CorA from Thermotoga maritima (Fig. 25a), solved by three different groups, reveals a homopentamer with a large cytoplasmic domain and a transmembrane domain consisting of 10 helices, two from each monomer (Eshaghi et al. Reference Eshaghi, Niegowski, Kohl, Martinez Molina, Lesley and Nordlund2006; Lunin et al. Reference Lunin, Dobrovetsky, Khutoreskaya, Zhang, Joachimiak, Doyle, Bochkarev, Maguire, Edwards and Koth2006; Payandeh & Pai, Reference Payandeh and Pai2006). All three structures probably represent a closed state of the channel. The amino terminal cytoplasmic domain resembles a funnel. Its structure represents a new fold made of a seven-stranded mixed β-sheet sandwiched between two set of α-helices (helices 1, 2, 3 and helices 4, 5, 6). Conserved residues from the relatively long helices 5 and 6 form an acidic ring. The interior of the cytoplasmic domain is lined by residues from helix 7 (the stalk helix), which extends into the membrane with a kink to form the pore helix TM1. A short periplasmic loop that contains the MPEL motif and is disordered in all structures connects TM1 and TM2. TM2 surrounds the pore helix and ends in the cytoplasm with a KKKKWL motif that lies perpendicular to the membrane and possibly interacts with lipids. Together these lysines and those from the stalk helix account for 30 positive charges, positioned above the acidic ring and just below the pore, giving rise to a region called the basic sphincter. Helix TM1 is twisted along the fivefold axis and tapers near the periplasmic surface (Fig. 25a). The pore forming helix TM1 is devoid of any charged residues, so the pore is predominantly lined by backbone carbonyls and amides along with a few serine and threonine hydroxyl groups. Only two residues (proline and phenylalanine) are conserved in the pore helix of the corA family. The pore helix is capped at the periplasmic end by asparagine from a GMN motif and at the cytoplasmic end by a conserved aspartate. The diameter of the pore varies from 6 to 2·5 Å, being smallest at the cytoplasmic end. Cationic hexamines that have similar size and shape to Mg2+ inhibit CorA, suggesting that a fully hydrated Mg2+ binds initially probably via the MPEL motif. The cation then has to travel across the bilayer losing its water on the way.

Fig. 25. Magnesium transport proteins (a) pentameric CorA (2IUB) and (b) dimeric MgtE (2YVX). Side and periplasmic views of CorA: Pentameric CorA is made up of a large cytoplasmic domain and a TM domain with 10 helices, 2 from each monomer (brown). The stalk helix (yellow) that connects the two domains might play a role in transmitting a signal to the TM domain. Helices 5 (cyan) and 6 (pink) and the rest of cytoplasmic domain (green) acts as a divalent cation sensor (bound magnesium ions shown as red spheres). The C-terminal extension of TM2 domain that has a KKKKWL motif (blue) forms part of a basic sphincter ring. There is no bound Mg2+ in the pore and this structure probably represents a closed state. Side and periplasmic views of MgtE: In contrast to CorA, MgtE is a homodimer with 10 TM helices (brown), 5 from each monomer. The cytoplasmic domain consists of a superhelical N domain (green) and a cystathionine-β-synthase domain (cyan). The two plug helices (yellow) connect the TM and cytoplasmic domains. Magnesium ions (red spheres) are found at the interfaces between domains. One Mg2+ ion has been modelled in the pore near a conserved aspartate but due to moderate resolution the exact nature of the bound ion or the co-ordination geometry cannot be determined. Although different in architecture, the cytoplasmic domains of CorA and MgtE both act as divalent cation sensors. The loss of ion co-ordination due to lower intracellular Mg2+ concentration results in movement of the stalk helix in CorA or the connecting helix in MgtE, which transmits the signal to the TM domain. It is important to note that the bound cations in the cytoplasmic domains are at the interfaces of the various subdomains in both proteins. Except for a single aspartate in MgtE, hydrophobic residues line the pores of both proteins.
In the present structures no density for Mg2+ is found in the pore. However, three constrictions are found in the pore that may play a role in selecting and conducting Mg2+. At the periplasmic end, the side chains of asparagines from each monomer face towards the pore and may toggle up and down to let the ions through. The second constriction made of threonine and methionine residues may play a role in dehydrating Mg2+. Towards the cytoplasmic end, hydrophobic leucine and methionine residues constrict the channel to ~2·5 Å. Leucine plugs the pore and may play a role in gating. In fact, replacement of leucine with smaller hydrophobic amino acids results in an increase in the open probability of the channel (Payandeh et al. Reference Payandeh, Li, Ramjeesingh, Poduch, Bear and Pai2008).
Below this leucine, there is a ring of conserved aspartate residues that might interact with incoming magnesium ions and deliver them to the cytoplasmic domain. The acidic and hydrophilic residues in helix 7 may function in attracting the cation as an electrostatic sink. However in the closed state, residues from neighbouring helices neutralise them. In the absence of an open state structure for CorA, structures of the cytoplasmic domain in the presence of divalent cations and biochemical analysis provide insights about sensing of intracellular Mg2+ concentration and signal transmission to the TM domain (Lunin et al. Reference Lunin, Dobrovetsky, Khutoreskaya, Zhang, Joachimiak, Doyle, Bochkarev, Maguire, Edwards and Koth2006; Payandeh & Pai, Reference Payandeh and Pai2006). In the closed state, the cytoplasmic domain of CorA has a basic ring followed by an acidic ring below the pore. Divalent cations at the interface of individual domains hold them tightly together. When the Mg2+ ion concentration is low in the cytoplasm, it is thought to lead to a loss of ion co-ordination in the cytoplasmic domain, followed by charge repulsion of the basic sphincter and the acidic ring resulting in opening of the channel, which is likely to involve a rotational and tilting movement of the stalk and other TM helices (Payandeh et al. Reference Payandeh, Li, Ramjeesingh, Poduch, Bear and Pai2008). The structures of CorA have been informative but do not provide answers to the questions of selectivity and conduction. A structure with a cation hexamine might provide a first clue about the entry of cation into the pore, followed by mutants selected to hold the channel in an open state, a scenario similar to MscS. Calcium, although found in lower concentrations in the cytoplasm binds to the same sites in the cytoplasmic domain as Mg2+, raising the question of how the competition between these ions is overcome.
MgtE
The crystal structure of MgtE from Thermus thermophilus at 3·5 Å reveals (Fig. 25b) a homodimer with each monomer contributing five helices, in straight contrast to the pentameric CorA channel that has the same number of helices altogether (Hattori et al. Reference Hattori, Tanaka, Fukai, Ishitani and Nureki2007). The N-terminal cytosolic domain of MgtE consists of a superhelical N-domain and tandemly repeated cystathionine-β-synthase (CBS) domain (Fig. 25b). The N-domain is a right-handed α-helical bundle made of ten helices. Duplicated CBS domains have been observed in other membrane proteins and are known to play a regulatory role (Ignoul & Eggermont, Reference Ignoul and Eggermont2005). Numerous acidic residues are clustered at the interface between the N and CBS domains. The cytoplasmic domain connects to the TM domain by a connecting helix (also called the plug helix) that lies perpendicular to the membrane (yellow in Fig. 25b).
The five TM helices are connected by loops L0 to L4. Loops 1 and 4 form short helices H1b and H4b. Most of the TM helices are kinked at glycine or proline residues. TM2 is surrounded by another four helices and does not traverse the membrane completely, thus accommodating the non-helical loop L1 to form the periplasmic entrance. The connecting helix interacts with TM2, TM5 and H4b through van der Waals interaction. Highly conserved acidic residues are clustered in connecting helix and in the loops of the TM helices. Residues from TM2 and TM5 with some contribution from TM1 form the ion conduction pathway. Except for a conserved aspartate in the middle of the bilayer, the pore is lined mostly by hydrophobic amino acids and their main chain carbonyls. The pore is ~30 Å long and 6 Å in diameter close to the cytosol where a number of hydrophilic residues compensate for the energetic cost of dehydration. The interaction of the connecting helix with the TM helices brings together the two TM5 helices and probably fixes the pore in a closed state.
At the present resolution it is not possible to assign densities to Mg2+ ions with confidence. One such density has been tentatively assigned as Mg2+ ion in the pore close to aspartate. Four other Mg2+ ions have been assigned at interfaces between the CBS and N domains and the connecting helix. These cytoplasmic Mg2+ ions can be assigned with some confidence based on the structure of the isolated cytoplasmic domain of MgtE in the presence or absence of Mg2+. These Mg2+ ions might stabilise the interaction between the N and CBS domains and fix the connecting helix for its interaction with TM2, TM5 and H4b. In a similar way to CorA, there has been speculation about the mechanism of gating based on the structure of the isolated cytoplasmic domain in the presence or absence of Mg2+ and by MD stimulation (Hattori et al. Reference Hattori, Tanaka, Fukai, Ishitani and Nureki2007; Ishitani et al. Reference Ishitani, Sugita, Dohmae, Furuya, Hattori and Nureki2008). In the presence of Mg2+ (as in the crystal structure), the CBS domain tightly dimerises, fixing the connecting helix to interact with the TM domain and thereby close the pore. In the absence of Mg2+, the dimer interface of the CBS domain loosens, allowing the possible rotation of the connecting helices so that they swing away from each other. This is probably followed by conformational change in the TM helices to open the pore. It is not surprising that two distinct families of Mg2+ transport proteins utilise similar mechanisms for sensing intracellular Mg2+ ions although their TM domain architecture is distinct. It is not clear whether MgtE acts as a channel or a transporter? In the absence of a functional assay but with structural similarity to the secondary active transporter YiiP (Lu & Fu, Reference Lu and Fu2007), it is tempting to believe that MgtE is also an active transporter.
Pore-forming toxins
A large family of proteins, secreted as soluble monomers can oligomerise to form pores on target cells. They are collectively called pore-forming toxins (PFT). They can be classified based on their α-helical or β-barrel structures as α-PFT or β-PFT. Both classes of protein are thought to undergo sequential steps from a soluble monomer to binding, oligomerisation and penetration into the target cell membrane. Many such toxins are secreted by bacterial pathogens but have also evolved in higher animals. Members of α-PFT include colicins and cytolysins (ClyA) of E. coli, cry toxins of Bacillus thuringiensis, exotoxin A of Pseudomonas aeruginosa and actinoporins of sea anemones. Anti-apoptotic protein Bcl-x has a similar structure to colicins but its relevance to pore formation remains to be elucidated (Anderluh & Lakey, Reference Anderluh and Lakey2008). β-PFT include α-hemolysin (α-HL) from Staphylococcus aureus, aerolysin from Aeromonas hydrophila, the anthrax protein antigen, the B. thuringiensis delta endotoxins and the cholesterol dependent cytolysins (Gouaux, Reference Gouaux1998; Iacovache et al. Reference Iacovache, Van der Goot and Pernot2008). Membrane attack complex proteins of the complement and perforin family have been predicted to form β-barrel pores. Thus, similar principles have been exploited by bacterial pathogens as well as higher eukaryotes either at the onset of infection or as a defence mechanism. It has been speculated that these toxins have different oligomeric states ranging from a monomer (colicin) to large assemblies (cholesterol dependent cytolysins). The absence of structures in a biologically relevant form limits the information on the oligomeric states but it is likely that many if not all these toxins must associate to form pores. Structures of ClyA and α-HL that have been crystallised and solved in the presence of detergent are representative of membrane inserted α- and β-PFTs, respectively (Song et al. Reference Song, Hobaugh, Shustak, Cheley, Bayley and Gouaux1996; Mueller et al. Reference Mueller, Grauschopf, Maier, Glockshuber and Ban2009).
In the presence of detergent, monomeric ClyA undergoes a massive structural transformation and oligomerises into a dodecameric pore (Fig. 26a). In its soluble form the ClyA monomer comprises four α-helices (A, B, C and F), a hydrophobic β-hairpin flanked by two short helices that form the head domain and the C-terminal helix G. A kink at a conserved proline in helix A separates it into helices A1 and A2. The head domain with its hydrophobic residues is thought to interact with the membrane initially and subsequent conformational change results in an all α-helical protein devoid of any β-sheets (Mueller et al. Reference Mueller, Grauschopf, Maier, Glockshuber and Ban2009). Translocation of helix A results in a conversion in the membrane form to a three-helix bundle and the region that comprised A2 refolds into an extension of helix B. The long helices B, C and F form the wall of the pore and the top is constricted by the N-terminus of helix A.
Fig. 26. Pore forming toxins a) ClyA (2WCD) b) α-HL (7AHL). (a) Side and top view of cytolysin A: in its membrane bound form, ClyA is a dodecameric α-helical pore-forming toxin. One subunit is highlighted to show the three-helix bundle. Helices B (green), C (yellow) and F (cyan) form the wall of the pore. Helix A (blue) and a short region of helix C comprise the membrane-inserted part. (b) Side and top view of α-hemolysin: α-HL is a β-barrel pore-forming toxin. The monomer of α-HL consists of the rim and amino latch domain (green) and a stem domain (blue) that forms the barrel. Each monomer contributes two β-strands to form a heptameric barrel.
α-HL is one of the best-studied toxins that oligomerises to form a heptamer in the membrane (Fig. 26b). Two β-strands from each soluble monomer associate in the membrane to form a large water-filled 14-TM β-barrel (Song et al. Reference Song, Hobaugh, Shustak, Cheley, Bayley and Gouaux1996). In the eukaryotic cell membrane α-HL pores release metabolites and ions, eventually leading to host cell lysis. Unlike ClyA, α-HL does not undergo such a large change in structure between its monomeric soluble form and that of membrane-inserted oligomer.
Key technologies
The enormous success of membrane protein structure determination has depended on a host of technical developments. No review on membrane protein structure would be complete without a brief mention of the key methods that have been developed to obtain purified proteins and determine their structures. We highlight here only a few aspects of expression and purification, as there are several recent papers that describe or review different approaches (Reeves et al. Reference Reeves, Kim and Khorana2002; Opekarova & Tanner, Reference Opekarova and Tanner2003; Kost et al. Reference Kost, Condreay and Jarvis2005; Sorensen & Mortensen, Reference Sorensen and Mortensen2005; Midgett & Madden, Reference Midgett and Madden2007; Junge et al. Reference Junge, Schneider, Reckel, Schwarz, Dotsch and Bernhard2008).
Membrane protein expression and purification
Expression systems have been developed to overcome some of the problems encountered during early attempts to overexpress membrane proteins. The need for specific membrane protein insertion machinery has often limited the capacity of a host cell to produce large quantities of correctly folded membrane proteins without toxic overload. Many over-expressed membrane proteins form inclusion bodies when expressed in E. coli, and even correctly inserted overexpressed membrane proteins can be trapped in the secretory pathway of eukaryotic cells in a non-functional state (Tate, Reference Tate2001). Because E. coli is most easily handled and has been used in screening the expression of large numbers of membrane proteins identified in prokaryotic genomes, it has been the most productive expression system so far. However, specific post-translational modifications such as glycosylation or the need for specific lipids have made the expression of eukaryotic proteins in E. coli more problematic, probably due to incorrect folding. The structures of only two eukaryotic membrane proteins produced in E. coli have been solved, both membrane associated proteins in eicosanoid and glutathione (MAPEG) metabolism (Ferguson et al. Reference Ferguson, Mckeever, Xu, Wisniewski, Miller, Yamin, Spencer, Chu, Ujjainwalla, Cunnighham, Evans and Becker2007; Jegerschold et al. Reference Jegerschold, Pawelzik, Purhonen, Bhakat, Gheorghe, Gyobu, Mitsuoka, Morgenstern, Jakobsson and Hebert2008; see section 8.3). More successful expression of functional eukaryotic membrane proteins has been obtained using yeast, insect and mammalian cells. Protein structures using material from eukaryotic expression systems have included the voltage-gated potassium channels (Long et al. 2005 a), G-protein-coupled receptors (Cherezov et al. Reference Cherezov, Rosenbaum, Hanson, Rasmussen, Thian, Kobilka, Choi, Kuhn, Weis, Kobilka and Stevens2007; Jaakola et al. Reference Jaakola, Griffith, Hanson, Cherezov, Chien, Lane, Ijzerman and Stevens2008; Warne et al. Reference Warne, Serrano-Vega, Baker, Moukhametzianov, Edwards, Henderson, Leslie, Tate and Schertler2008), aquaporins (Hiroaki et al. Reference Hiroaki, Tani, Kamegawa, Gyobu, Nishikawa, Suzuki, Walz, Sasaki, Mitsuoka, Kimura, Mizoguchi and Fujiyoshi2006; Tornroth-Horsefield et al. Reference Tornroth-Horsefield, Wang, Hedfalk, Johanson, Karlsson, Tajkhorshid, Neutze and Kjellbom2006; Horsefield et al. Reference Horsefield, Norden, Fellert, Backmark, Tornroth-Horsefield, Terwisscha van Scheltinga, Kvassman, Kjellbom, Johanson and Neutze2008), P-glycoprotein (Aller et al. Reference Aller, Yu, Ward, Weng, Chittaboina, Zhuo, Harrell, Trinh, Zhang, Urbatsch and Chang2009) and P-type ATPase (Pedersen et al. Reference Pedersen, Buch-Pedersen, Morth, Palmgren and Nissen2007). Most of these have used yeast or baculovirus mediated expression in insect cells. The only structure of an over-expressed membrane protein from mammalian cells (COS cells) is that of recombinant rhodopsin, a success presumably aided by prior biochemical and crystallisation knowledge and the use of micro-crystallography (Standfuss et al. Reference Standfuss, Xie, Edwards, Burghammer, Oprian and Schertler2007). To overcome the toxic effects during over-expression, cell-free translation systems have been advocated (Junge et al. Reference Junge, Schneider, Reckel, Schwarz, Dotsch and Bernhard2008). Although there are numerous reports describing the production of milligram amounts of functional protein, only one structure (of EmrE) has been solved from material obtained from a cell-free translation system (Chen et al. Reference Chen, Pornillos, Lieu, Ma, Chen and Chang2007).
The stability of membrane proteins is often dramatically decreased upon solubilisation from the membrane, even in relatively mild detergents such as dodecylmaltoside. Fast purification strategies involving one or two steps are therefore advantageous and affinity tags have provided the most successful method to achieve rapid high enrichment in a single step. Selective enrichment of functional protein through the use of a ligand affinity column where possible is also a great advantage as this selectively purifies only correctly folded membrane proteins. The choice of detergent crucially influences the functional state of protein and the ability to obtain crystals that diffract well, so it is critical to screen a large number of detergents to find the best ones. Conventionally, the behaviour of membrane proteins in particular detergents has been tested by purification, especially size-exclusion chromatography, followed by functional assays and crystallisation. However, the ability to make fluorescent fusion proteins, in combination with size-exclusion chromatography, now allows easy and rapid screening of detergents for a particular protein without the need for purification (Kawate & Gouaux, Reference Kawate and Gouaux2006).
One of the major obstacles with many membrane proteins is the presence of multiple conformational states in solution. A particular conformational state can often be selected by use of inhibitors or substrate analogues as described for many transporters (Pebay-Peyroula et al. Reference Pebay-Peyroula, Dahout-Gonzalez, Kahn, Trezeguet, Lauquin and Brandolin2003; Locher, Reference Locher2008; Toyoshima, Reference Toyoshima2008). A relatively new approach is to identify mutations that promote both a particular conformational state and improved stability in comparison with the wild-type protein, named conformational thermostabilisation (Serrano-Vega et al. Reference Serrano-Vega, Magnani, Shibata and Tate2008). One of the earliest examples of a single mutation that decreased conformational flexibility while also increasing stability was found in LacY (Smirnova & Kaback, Reference Smirnova and Kaback2003). This application of systematic mutagenesis coupled with an appropriate ligand-binding assay has resulted in stable GPCRs which otherwise have a limited half-life in detergent micelles (Fig. 27) (Magnani et al. Reference Magnani, Shibata, Serrano-Vega and Tate2008; Serrano-Vega et al. Reference Serrano-Vega, Magnani, Shibata and Tate2008; Shibata et al. Reference Shibata, White, Serrano-Vega, Magnani, Aloia, Grisshammer and Tate2009). The relative simplicity of this approach, though it can be laborious to execute, makes it applicable to any membrane protein with a binding assay.
Fig. 27. Some membrane protein structures are partially perturbed in detergent micelles. Most membrane proteins have been crystallised in the presence of detergent, which is not a native environment. There are a few examples where the presence of detergents and strenuous efforts in crystallisation have caused non-native structures to form well diffracting crystals. (a) The small multi-drug transporter EmrE normally exists as an antiparallel dimer in the membrane as well as in detergent micelles (Fig. 14 c). However, in one of the crystal forms obtained by 3D crystallisation, TM helices 1 to 3 from each monomer (blue and yellow) pack against each other to form a dimer, similar to that found in the native structure obtained in presence of substrate or in a lipid bilayer (Ubarretxena-Belandia et al. Reference Ubarretxena-Belandia, Baldwin, Schuldiner and Tate2003; Chen et al. Reference Chen, Pornillos, Lieu, Ma, Chen and Chang2007; Fig. 14 c). However the TM 4 helices (red) are directed entirely away from the dimer core in all the monomers of the asymmetric unit. It is possible that the low pH used for crystallisation promotes disruption of the normal helix bundle and that contacts made by TM4 results in a well-ordered lattice. (b) Four independent molecules of β1 adrenergic receptor are observed in one crystal form. In two of these molecules, the N-terminus of TM helix 1, representing about 10 amino acids, adopts a significantly different conformation from the structure of the two other molecules that represent the antagonist-bound state. The figure shows an overlay of molecules A and B (aquamarine), with the 60°-kinked conformation of the N-terminus of TM1 in molecule A coloured in red and the correct, non-kinked conformation of TM1 in molecule B in aquamarine. TM1 in GPCRs is the most distant helix from both the ligand binding and the G-protein binding sites and such distortion is certainly a result of crystallisation.
Three-dimensional crystallisation and X-ray crystallography
Once a membrane protein has been purified in a functional, stable and monodisperse state, 3D crystals may be relatively easily obtained but membrane protein crystals are often very small and disordered. The properties of the detergent and its final concentration are especially important (Ostermeier & Michel, Reference Ostermeier and Michel1997; Kuhlbrandt, Reference Kuhlbrandt1988; Prive, Reference Prive2007). It is often necessary to screen a large number of crystallisation conditions and make many trips to synchrotrons.
New strategies developed over the last 10 years increase the probability of obtaining good diffracting crystals. Michel (1983) originally distinguished type I (stacks of 2D layers growing in the third dimension) from type II (involving contacts principally between hydrophilic parts of the protein) crystals. With conventional vapour diffusion methods, type II crystals are more commonly observed. Since detergents form a belt around solubilised membrane proteins, detergents that form small micelles have been most successful in obtaining high-resolution structures with type II crystals. However, many proteins are stable only in detergents with large micelles, so different approaches are required in such cases. It is the size of the protein relative to the size of the detergent micelle that is the key factor. Dodecylmaltoside has been used successfully to crystallise larger membrane protein complexes, but membrane proteins with only small hydrophilic domains protruding from their hydrophobic lipid-embedded regions need detergents with small micelles and possibly other modifications. These include the use of antibodies or fusion proteins to increase the non-detergent surface area of the protein:detergent micelle, or modulating the property of detergents by addition of small molecules, or using mixed detergents. Since membrane proteins have evolved in lipid bilayers and not in detergent micelles it is also desirable to determine the structure in a more native environment (Fig. 27). This can be achieved either by 2D crystallisation (see section 12.3), the use of bicelles which are lipid-enriched detergent micelles (Faham & Bowie, Reference Faham and Bowie2002), lipidic cubic phase with appropriate matrices (Caffrey, Reference Caffrey2003) or by simple addition of specific lipids during purification and crystallisation (Guan et al. Reference Guan, Mirza, Verner, Iwata and Kaback2007). Success with lipidic cubic phase crystallisation started with proteins from the bacteriorhodopsin family but has been extended to other membrane proteins including GPCRs (e.g. Cherezov et al. Reference Cherezov, Rosenbaum, Hanson, Rasmussen, Thian, Kobilka, Choi, Kuhn, Weis, Kobilka and Stevens2007). The availability of microfocus beams (1–10 μm diameter) allows scanning along the crystal to find particular areas that are better ordered, as well as complete 3D data collection using tiny crystals.
Electron cryomicroscopy
Six membrane protein structures have been determined to better than 3·5-Å resolution by electron cryomicroscopy. These are bacteriorhodopsin (Henderson & Unwin, Reference Henderson and Unwin1975; Henderson et al. Reference Henderson, Baldwin, Ceska, Zemlin, Beckmann and Downing1990), LHCII from green plants (Kühlbrandt et al. 1994 shown in Fig. 28a), aquaporins AQP1 (Murata et al. Reference Murata, Mitsuoka, Hirai, Walz, Agre, Heymann, Engel and Fujiyoshi2000) and AQP0 (Gonen et al. Reference Gonen, Sliz, Kistler, Cheng and Walz2004b), glutathione S-transferase (Holm et al. Reference Holm, Bhakat, Jegerschold, Gyobu, Mitsuoka, Fujiyoshi, Morgenstern and Hebert2006) and prostaglandin E synthetase (Jegerschold et al. Reference Jegerschold, Pawelzik, Purhonen, Bhakat, Gheorghe, Gyobu, Mitsuoka, Morgenstern, Jakobsson and Hebert2008). The highest resolution obtained to date is 1·9 Å for AQP0 (Gonen et al. Reference Gonen, Sliz, Kistler, Cheng and Walz2004b). Numerous other structures have been determined to 5- to 9-Å resolution, which allows the architecture of α-helical membrane proteins to be determined (reviewed by Henderson, 2004). These low-resolution analyses are relatively quick and frequently very informative about secondary structure. By using other biochemical and evolutionary data together with hydrophobic moment calculations, these lower resolution maps can often be used to derive good working models of the structure, e.g. EmrE (Fleishman et al. Reference Fleishman, Harrington, Enosh, Halperin, Tate and Ben-Tal2006).
Fig. 28. Contributions from electron cryomicroscopy. (a) The structure of trimeric light-harvesting complex LHCII from pea (2W7B), determined by electron crystallography of 2D crystals, revealed a three-TM helical bundle and cofactors (chlorophyll in green and lutenes in salmon). (b) One example of how a low resolution 3D structure of a complete membrane protein complex, F1Fo-ATPase from bovine mitochondria, determined by single particle electron cryomicroscopy, can help to create a framework for interpreting the substructures of different parts of the whole complex determined by X-ray crystallography or NMR spectroscopy. Figure from Dickson et al. (Reference Dickson, Silvester, Fearnley, Leslie and Walker2006) with permission.
Structures determined using one technique, can also be used to help solve or validate structures determined by another technique. It is notable that the X-ray crystallographic structures of bacteriorhodopsin (section 3.1.2) have all been solved directly or indirectly from the original structures that were experimentally phased from electron microscope images. The clear structural relationship between the acetylcholine receptor, determined by electron microscopy, and the bacterial homopentameric GLIC and ELIC channels, determined by X-ray crystallography, both validates the relevance of the bacterial structures to the pentameric ligand-gated channel family in general and confirms the correctness of the EM work (see section 2.2).
Electron microscopy also has a valuable role to play in other special cases. One of these is the case of intransigent structures. It is notable that the structure of eukaryotic αβ-tubulin (Nogales et al. Reference Nogales, Wolf and Downing1997) has been determined only by electron crystallography of 2D zinc-induced sheets. Another role is in the investigation of functional changes in conformation that might disrupt a 3D but not a 2D lattice. The large structural change found in bacteriorhodopsin at the mid-point of the photocycle in the purple membrane of halobacteria appears either to be prevented by crystal packing forces in 3D crystals, or to cause disorder so that the nature of the structural change so far could not be investigated by X-ray diffraction. Electron microscopy has been the only method that has been successful in describing this key structural change at high resolution (Subramaniam & Henderson, Reference Subramaniam and Henderson2000).
Finally, the growing power of single particle electron cryomicroscopy is allowing the structures of molecules or molecular complexes of interest to be obtained without making crystals. In this method, the structure is determined by computational alignment of different views of the same structure after embedding them in a thin film of amorphous ice. The method still depends on the averaging of identical structures, but these no longer need to be crystallised. A good example of this is shown in Fig. 28b (Rubinstein et al. Reference Rubinstein, Walker and Henderson2003). In this case, a relatively low-resolution structure for the F1Fo-ATPase from bovine mitochondria has allowed many other higher resolution structures for individual protein components or subcomplexes to be docked into the overall structure of the whole complex, producing valuable insights.
NMR spectroscopy
Solution NMR, as a method for protein and nucleic acid structure determination, has had considerable impact, especially for smaller proteins or molecules with partially disordered regions that have inhibited crystallisation attempts. However, NMR of membrane proteins has been difficult. First, the surrounding detergent micelle frequently doubles the effective size of the protein so that the tumbling rate is lower and therefore the width of the spectral peaks is larger. The need for sharper spectral peaks is usually tackled by using an elevated temperature to increase tumbling rate, but this is hampered for membrane proteins by their thermal instability, particularly in the smaller detergents that make smaller micelles. Second the chemical shifts for α-helical residues is concentrated in a smaller region of the spectrum, so that peaks often overlap, increasing even more the need for sharper peaks. In contrast β-barrel structures have proved easier to solve by NMR, because of the greater spectral dispersion.
As a result, there have been only a few structures determined by NMR spectroscopy (see Fig. 29 for some examples). One of the most interesting is the trimeric membrane enzyme, diacylglycerol kinase (Fig. 29c), in which the three TM helices in each of the three polypeptide chains make more intimate contact with helices in the other subunits than they do with helices in the same subunit (van Horn et al. 2009). In this case, it is notable that, despite great efforts to make well-ordered crystals, X-ray crystallographic approaches have not yet been successful, whereas NMR has. A promising recent success has also been the complete assignment of the backbone spectrum obtained from the halobacterial membrane protein, sR-II, which has seven TM helices, in micelles of the detergent DHPC at 50°C (Gautier et al. 2008).
Fig. 29. NMR structures of membrane proteins (a) OmpX (1Q9F), (b) OmpG (2JQY), (c) diacyl glycerol (DAG) kinase (2KDC) and (d) phospholamban (1ZLL). All membrane protein structures so far determined by NMR of detergent solubilised proteins in solution are relatively small structures because the large size of the protein-detergent micelle complex broadens the NMR spectrum. Two representative NMR structures of the β-barrel membrane proteins OmpX, an eight-stranded β-barrel, and OmpG, a 14-stranded β-barrel are shown in (a) and (b), respectively. The largest β-barrel membrane protein structure determined by NMR is the mitochondrial porin VDAC (see section 9 and Fig. 24d). The structure of diacyl glycerol kinase (c) has so far been determined only by NMR spectroscopy. It reveals a trimer, each subunit having three TM helices that all make extensive intersubunit contacts. Phospholamban is a pentameric channel associated with the sarcoplasmic reticulum Ca2+-ATPase, SERCA. These two represent the α-helical membrane protein structures determined by NMR.
NMR has also helped greatly in characterising the detailed energetics of helix-helix packing in simple systems, such as found in the glycophorin A dimer, the first NMR structure of a membrane protein, in which the helix packing of the GxxxG motif generates an unusual but very stable left-handed transmembrane helix dimer (MacKenzie et al. Reference Mackenzie, Prestegard and Engelman1997). Finally, solid state NMR may be developed into a more powerful technique in future, but at present the amount of information obtainable is limited to analysis of the conformation of specifically labelled parts of the structure such as the peptide ligand bound to a peptide receptor (Luca et al. 2003) or retinal bound to rhodopsin (Ahuja et al. Reference Ahuja, Crocker, Eilers, Hornak, Hirshfeld, Ziliox, Syrett, Reeves, Khorana, Sheves and Smith2009).
Future prospects and challenges
In our initial overview, we argued that membrane protein structure determination appeared to have progressed through three phases, first for natural abundance proteins, then those that were selected from a behavioural screen of expressed genes identified from bacterial whole genome sequences and, finally, those where substantial further work was needed to increase the stability of the proteins once removed from their lipid environment. Looking to the future, what new trends can we anticipate?
From the range of structural patterns found in membrane proteins so far, the preponderance of α-helical bundles and β-barrel structures provides one great simplification. Both these classes of structure show a clear distinction between the simple closed secondary structure geometry in transmembrane regions and the much more convoluted surfaces outside the bilayer region that includes loops, turns and other complex folds that are typically found in soluble, non-membrane proteins. Occasional mixed structures are observed (e.g. FepA in Fig. 24b, or NalP in Fig. 24c), but why have no αβ-barrel structures been observed, such as those first observed in triose phosphate isomerase, the so-called TIM-barrel, in which an eight-stranded β-barrel is surrounded by eight α-helices? Why are there no membrane protein structures with complete β-barrels surrounded by a small number of TM α-helices? Such structures, once formed, should be stable in the lipid bilayer. Perhaps the answer is that the distribution of hydrophobic and hydrophilic residues that would be required for stable insertion makes the polypeptide aggregate in solution or get stuck inside the ribosome. Such mixed αβ structures might still be found in membrane protein complexes such as the mitochondrial import apparatus (Mokranjac & Neupert, Reference Mokranjac and Neupert2009). Another structural feature that recurs is the existence of breaks in TM α-helices that contribute to a functional need, for example by providing ion binding sites at exposed backbone carbonyl groups in the short non-helical regions. Glycine and proline residues in TM helices also introduce kinks and bends that are important for gating or signal transduction. These are found in many membrane proteins that function as channels or transporters.
One future trend will be towards more accurate calculations of folding energy. Theoretical calculations based on energy minimisation, helped by molecular dynamics, are steadily improving (Qian et al. Reference Qian, Raman, Das, Bradley, Mccoy, Read and Baker2007). It may soon be possible to extrapolate known structures in the computer in a reliable way.
There are only a finite number of structures in biology. Current estimates suggest about 25 000 human genes of which about one third are membrane proteins. The number of unique membrane protein folds may thus be not much more than a few hundred, so we should have a good idea of the structures of most of them within the next 10–20 years. Homologous structures could then be modelled and refined by energy minimisation. At that point, there will still be a need to determine the structures of complexes, to describe structural changes that are so finely balanced that even the most accurate energy calculations will be unable to cope, to investigate mechanisms, and above all to design drugs that have practical value in treatment of disease or infection or as tools to investigate biological function. There will be a gradual transition from discovery to invention, a trend that is already well advanced in many laboratories. Two recent examples of the inventive use of structural knowledge are the construction of novel α-hemolysin (α-HL) nanopores and the coupling of ion channels to receptors. Due to its simplicity and ability to tolerate mutations, α-HL has become a prototype for biotechnological engineering in which the pore is reconfigured to become a sensor (Gu et al. Reference Gu, Braha, Conlan, Cheley and Bayley1999; Maglia et al. Reference Maglia, Restrepo, Mikhailova and Bayley2008; Stoddart et al. Reference Stoddart, Heron, Mikhailova, Maglia and Bayley2009). This includes the ability to distinguish the passage of the different nucleotides in DNA, which in future might make it possible to speed up the sequencing of whole genomes (Stoddart et al. Reference Stoddart, Heron, Mikhailova, Maglia and Bayley2009). In another novel experiment, two different G-protein-coupled receptors have been fused to an ion channel and shown to produce increases or decreases in membrane current on addition of the receptor agonist (Moreau et al. Reference Moreau, Dupuis, Revilloud, Arumugam and Vivaudou2008).
Finally, there are bound to be unanticipated surprises. History is littered with statements that imply everything will soon be known. Vannevar Bush's analysis of science as an endless frontier is just as true for membrane protein structure as it is in every other area (Bush, Reference Bush1945).
Acknowledgements
We would like to thank Declan Doyle, Bob Gennis, Werner Kuehlbrandt, Edmund Kunji, Andrew Leslie, Greg McMullan, Fiona Marshall, Jim Naismith, Chris Tate, Nigel Unwin and Wolfram Welte for their valued suggestions and critical reading of manuscript.
We also thank Joanna Westmoreland for help with figures. Vinothkumar was a recipent of a Marie-Curie Intra-European Fellowship (FP6) and an MRC career development fellowship.






























 Open access
Open access