
Contents
Articles
Effects of Long-Time Current Annealing to the Hysteresis in CVD Graphene on SiO2
-
- Published online by Cambridge University Press:
- 02 October 2019, pp. 3319-3326
-
- Article
- Export citation
Synthesis and Characterization of Laser Induced Graphene (LIG) and laser Reduced Graphene Oxide (lrGO) by using a pulsed CO2 laser
-
- Published online by Cambridge University Press:
- 23 December 2019, pp. 3327-3335
-
- Article
- Export citation
The role of surface morphology on nucleation density limitation during the CVD growth of graphene and the factors influencing graphene wrinkle formation
-
- Published online by Cambridge University Press:
- 28 January 2020, pp. 3337-3345
-
- Article
- Export citation
Influence of microwave photo-excitation on the transport properties of the high mobility GaAs/AlGaAs 2D electron system
-
- Published online by Cambridge University Press:
- 21 January 2020, pp. 3347-3352
-
- Article
- Export citation
Ultrathin molecule-based magnetic conductors: A step towards flexible electronics
-
- Published online by Cambridge University Press:
- 16 December 2019, pp. 3353-3364
-
- Article
- Export citation
Electronic structure study of new family of high-Tc Fe-superconductors based on BaFe2As2 in presence of dopants Rh and Pd.
-
- Published online by Cambridge University Press:
- 04 November 2019, pp. 3365-3372
-
- Article
- Export citation
Multi-Objective Optimization of the Hot Rolling Scheduling of Steel Using a Genetic Algorithm
-
- Published online by Cambridge University Press:
- 19 November 2019, pp. 3373-3380
-
- Article
- Export citation
Impact of Molecular Dynamics Simulations on Research and Development of Semiconductor Materials
-
- Published online by Cambridge University Press:
- 23 September 2019, pp. 3381-3398
-
- Article
- Export citation
-
Atomic scale defects critically limit performance of semiconductor materials. To improve materials, defect effects and defect formation mechanisms must be understood. In this paper, we demonstrate multiple examples where molecular dynamics simulations have effectively addressed these issues that were not well addressed in prior experiments. In the first case, we report our recent progress on modelling graphene growth, where we found that defects in graphene are created around periphery of islands throughout graphene growth, not just in regions where graphene islands impinge as believed previously. In the second case, we report our recent progress on modelling TlBr, where we discovered that under an electric field, edge dislocations in TlBr migrate in both slip and climb directions. The climb motion ejects extensive vacancies that can cause the rapid aging of the material seen in experiments. In the third case, we discovered that the growth of InGaN films on (0001) surfaces suffers from a serious polymorphism problem that creates enormous amounts of defects. Growth on (
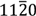 $11\bar{2}0$) surfaces, on the other hand, results in single crystalline wurtzite films without any of these defects. In the fourth case, we first used simulations to derive dislocation energies that do not possess any noticeable statistical errors, and then used these error-free methods to discover possible misuse of misfit dislocation theory in past thin film studies. Finally, we highlight the significance of molecular dynamics simulations in reducing defects in the design space of nanostructures.
$11\bar{2}0$) surfaces, on the other hand, results in single crystalline wurtzite films without any of these defects. In the fourth case, we first used simulations to derive dislocation energies that do not possess any noticeable statistical errors, and then used these error-free methods to discover possible misuse of misfit dislocation theory in past thin film studies. Finally, we highlight the significance of molecular dynamics simulations in reducing defects in the design space of nanostructures.
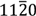
Adsorption mechanism of acid orange 7 on photocatalytic materials based on TiO2
-
- Published online by Cambridge University Press:
- 23 December 2019, pp. 3399-3405
-
- Article
- Export citation
PHOTOCATALYTIC PERFORMANCE OF ZnO/N-rGO FOR LIGNIN DEGRADATION UNDER VIS LIGHT ENERGY
-
- Published online by Cambridge University Press:
- 04 November 2019, pp. 3407-3415
-
- Article
- Export citation
Polymer-cellulosic-clay and polymer-cellulosic-zeolitic biocomposites for the removal of methylene blue in aqueous solution
-
- Published online by Cambridge University Press:
- 19 November 2019, pp. 3417-3421
-
- Article
- Export citation
Changing width bandgap of TiO2 nanoparticles incorporating GO
-
- Published online by Cambridge University Press:
- 10 January 2020, pp. 3423-3431
-
- Article
- Export citation
Physicochemical characterization of Pt and Ir particles deposited on Ce(1-x)Ru(x)O2 solid-solutions for CO oxidation
-
- Published online by Cambridge University Press:
- 18 November 2019, pp. 3433-3440
-
- Article
- Export citation
Front Cover (OFC, IFC) and matter
ADV volume 4 issue 61-62 Cover and Front matter
-
- Published online by Cambridge University Press:
- 11 February 2020, pp. f1-f4
-
- Article
-
- You have access
- Export citation
Back matter (Indexes)
ADV volume 4 issue 61-62 Author and Subject Indexes
-
- Published online by Cambridge University Press:
- 11 February 2020, pp. b1-b3
-
- Article
- Export citation