



1. Introduction
Using the premise that effective variants are in linkage disequilibrium (LD) with common polymorphisms and haplotypes, linkage and association studies have identified genes involved in the development of traits and pathologies. Upon their identification, the regions flanking associated markers are sequenced to find the linked, penetrant variant. However, rare variants are often not detectable using LD-based methods. This problem has been alleviated by recent advances in next-generation sequencing (NGS), and the detection of highly penetrant rare variants associated with disease has reduced the heritability gap for such diseases as autism, Crohn's disease and osteoporosis (Kosmicki et al., Reference Kosmicki, Churchhouse, Rivas and Neale2016; Bomba, et al. Reference Bomba, Walter and Soranzo2017). Despite these advances, for most traits and complex disorders the underlying genes and variants remain elusive.
Recessive disorders are caused by mutations in both copies of a gene. The mutations may be homozygous, that is, identical or compound heterozygous. Compound heterozygous (CH) variants are two different variants in a gene on opposite alleles of a chromosome and it is speculated that CH mutations account for many recessive diseases (Li et al., Reference Li, Vinckenbosch, Tian, Huerta-Sanchez, Jiang, Jiang, Albrechtsen, Andersen, Cao, Korneliussen, Grarup, Guo, Hellman, Jin, Li, Liu, Liu, Sparsø, Tang, Wu, Wu, Yu, Zheng, Astrup, Bolund, Holmkvist, Jørgensen, Kristiansen, Schmitz, Schwartz, Zhang, Li, Yang, Wang, Hansen, Pedersen, Nielsen and Wang2010; Sanjak et al., Reference Sanjak, Long and Thornton2017). Lack of detection of CH variants may explain a significant portion of missing heritability for all phenotypes (Li et al., Reference Li, Vinckenbosch, Tian, Huerta-Sanchez, Jiang, Jiang, Albrechtsen, Andersen, Cao, Korneliussen, Grarup, Guo, Hellman, Jin, Li, Liu, Liu, Sparsø, Tang, Wu, Wu, Yu, Zheng, Astrup, Bolund, Holmkvist, Jørgensen, Kristiansen, Schmitz, Schwartz, Zhang, Li, Yang, Wang, Hansen, Pedersen, Nielsen and Wang2010; Zhong et al., Reference Zhong, Karssen, Kayser and Liu2016; Sanjak et al., Reference Sanjak, Long and Thornton2017). Association studies using polymorphisms are LD-based and recent association studies using rare variants compare total variant burden between cases and controls to account for the contributions of multiple alleles at a locus to phenotype. Importantly, because LD-based studies require recessive variants to be on the same genetic background and total variant burden analyses are not allele specific, neither discerns between dominant and recessive models of inheritance.
Burden tests may account for compound heterozygosity if the variants are allocated to one of the two alleles for a gene, that is, phased. Relatively common variants may be phased assuming linkage to surrounding haplotypes; in families, rare variants are phased using parental genotypes. Once variants are phased, it may be determined if an individual's variants are on different chromosomes, and burden tests that aggregate using an indicator function (i.e. presence of qualifying variants) may assess enrichment for recessive variants.
Here we provide a publicly available tool, VarCount, that is user-friendly and effective for researchers seeking to quantify the presence or absence of a variant or variants in a gene at the individual level. VarCount is useful for the quantification of heterozygous, homozygous or CH variants per sample. We used VarCount to query the Epi4k (Epi4k Consortium et al., Reference Allen, Berkovic, Cossette, Delanty, Dlugos, Eichler, Epstein, Glauser, Goldstein, Han, Heinzen, Hitomi, Howell, Johnson, Kuzniecky, Lowenstein, Lu, Madou, Marson, Mefford, Esmaeeli Nieh, O'Brien, Ottman, Petrovski, Poduri, Ruzzo, Scheffer, Sherr, Yuskaitis, Abou-Khalil, Alldredge, Bautista, Berkovic, Boro, Cascino, Consalvo, Crumrine, Devinsky, Dlugos, Epstein, Fiol, Fountain, French, Friedman, Geller, Glauser, Glynn, Haut, Hayward, Helmers, Joshi, Kanner, Kirsch, Knowlton, Kossoff, Kuperman, Kuzniecky, Lowenstein, McGuire, Motika, Novotny, Ottman, Paolicchi, Parent, Park, Poduri, Scheffer, Shellhaas, Sherr, Shih, Singh, Sirven, Smith, Sullivan, Lin Thio, Venkat, Vining, Von Allmen, Weisenberg, Widdess-Walsh and Winawer2013) dataset for rare homozygous and CH variants and found enrichment for rare, CH variants in six genes, including three involved in neuronal development or growth (PRTG, TNC and MACF1). The variants in the 1000 Genomes Project database are now phased (1000 Genomes Project Consortium et al., Reference Abecasis, Altshuler, Auton, Brooks, Durbin, Gibbs, Hurles and McVean2010; Reference Abecasis, Auton, Brooks, DePristo, Durbin, Handsaker, Kang, Marth and McVean2012; Reference Auton, Brooks, Durbin, Garrison, Kang, Korbel, Marchini, McCarthy, McVean and Abecasis2015), and so genes may be queried for in trans combinations of variants. The Epi4k enrichment was identified in comparison to the 1000 Genomes Project participants combining all ancestries and considering only individuals of European ancestry.
2. Materials and methods
(i) Processing of Epi4k vcf files
The Epi4k data (Epi4k Consortium et al., Reference Allen, Berkovic, Cossette, Delanty, Dlugos, Eichler, Epstein, Glauser, Goldstein, Han, Heinzen, Hitomi, Howell, Johnson, Kuzniecky, Lowenstein, Lu, Madou, Marson, Mefford, Esmaeeli Nieh, O'Brien, Ottman, Petrovski, Poduri, Ruzzo, Scheffer, Sherr, Yuskaitis, Abou-Khalil, Alldredge, Bautista, Berkovic, Boro, Cascino, Consalvo, Crumrine, Devinsky, Dlugos, Epstein, Fiol, Fountain, French, Friedman, Geller, Glauser, Glynn, Haut, Hayward, Helmers, Joshi, Kanner, Kirsch, Knowlton, Kossoff, Kuperman, Kuzniecky, Lowenstein, McGuire, Motika, Novotny, Ottman, Paolicchi, Parent, Park, Poduri, Scheffer, Shellhaas, Sherr, Shih, Singh, Sirven, Smith, Sullivan, Lin Thio, Venkat, Vining, Von Allmen, Weisenberg, Widdess-Walsh and Winawer2013) were accessed by permission via the Database of Genotypes and Phenotypes (dbGaP Study Accession, phs000653.v2.p1). Individual vcf files were combined using the CombineVariants function in GATK (McKenna et al., Reference McKenna, Hanna, Banks, Sivachenko, Cibulskis, Kernytsky, Garimella, Altshuler, Gabriel, Daly and DePristo2010). The vcf files were then annotated with minor allele frequencies (MAFs) from EVS (Exome Variant Server, NHLBI GO Exome Sequencing Project [ESP], Seattle, WA [URL: http://evs.gs.washington.edu/ EVS/]), 1000 Genomes Project and ExAC (Lek et al., Reference Lek, Karczewski, Minikel, Samocha, Banks, Fennell, O'Donnell-Luria, Ware, Hill, Cummings, Tukiainen, Birnbaum, Kosmicki, Duncan, Estrada, Zhao, Zou, Pierce-Hoffman, Cooper, DePristo, Do, Flannick, Fromer, Gauthier, Goldstein, Gupta, Howrigan, Kiezun, Kurki, Moonshine, Natarajan, Orozco, Peloso, Poplin, Rivas, Ruano-Rubio, Ruderfer, Shakir, Stenson, Stevens, Thomas, Tiao, Tusie-Luna, Weisburd, Won, Yu, Altshuler, Ardissino, Boehnke, Danesh, Roberto, Florez, Gabriel, Getz, Hultman, Kathiresan, Laakso, McCarroll, McCarthy, McGovern, McPherson, Neale, Palotie, Purcell, Saleheen, Scharf, Sklar, Patrick, Tuomilehto, Watkins, Wilson, Daly and MacArthur2015), and with information regarding the effect of each variant using SNPSift/SNPEff (Cingolani et al., Reference Cingolani, Platts, Wang le, Coon, Nguyen, Wang, Land, Lu and Ruden2012). The databases used for annotation were dbNSFP2.9 (for MAF and CADD score) and GRCh37.75 for protein effect prediction. SNPSift was used to remove any variants not inducing a protein-changing event (not ‘HIGH’ or ‘MODERATE’ impact) based on SNPEff annotation – this includes missense, nonsense, splice-site and insertion/deletion variants. Variants with quality flags and multiallelic variants, that is, those with more than two known nucleotide values, were also removed. Variants remaining after filtering were cross-referenced with the 1000 Genomes Project variants from the same MAF threshold to ensure that any variants removed from one dataset were removed from the other. The annotated vcf was used as input for VarCount. Ancestry for each exome was determined using LASER (Wang et al., Reference Wang, Zhan, Bragg-Gresham, Kang, Stambolian, Chew, Branham, Heckenlively, Fulton, Wilson, Mardis, Lin, Swaroop, Zollner and Abecasis2014) and this information was input to VarCount via the SampleInfo.txt file. Ancestry and phenotype information for each proband are described in Supplementary Table 1. In addition to the annotated vcf file, the parameters.txt and subjectinfo.txt (containing sex and ancestry information) were used as input. Within the parameters file, the following qualifications were selected: (1) counting at the transcript (rather than gene) level, (2) protein-changing effects, (3) MAF threshold of either 0.005 or 0.01, (4) all within-dataset and annotated (1000 Genomes Project, ExAC and EVS) MAFs, and (5) either CH or homozygous variants. Analyses were run separately for the two MAFs and using all Epi4k probands (264) and only those of European ancestry (207). Because the variants were not phased, VarCount was used to query the vcf file for individuals with two or more variants in each transcript. The output, a list of counts for each transcript was then used to query the parental vcf files for genotype information to determine which sets of variants composed in trans combinations of variants. Final counts were determined using parental genotype information. Custom python scripts were used to query for parental genotypes and to count true compound heterozygotes or homozygotes. De novo variants were excluded in the determination of true in trans variants.
(ii) Processing of 1000 Genomes Project vcf files
Vcf files for the 2504 participants in the 1000 Genomes Project (1000 Genomes Project et al., Reference Auton, Brooks, Durbin, Garrison, Kang, Korbel, Marchini, McCarthy, McVean and Abecasis2015) were downloaded by chromosome from the 1000 Genomes Project ftpsite. To reduce input file size, the genomic regions for the hg19 mRNA transcripts were downloaded via UCSC's Table Browser and used to remove noncoding regions from the vcf files. Including all exons from UCSC allowed for a more conservative analysis, given that the Epi4k data were sequenced using various exome captures, which are not inclusive of all possible exons. The variants were annotated and filtered via the same steps as the Epi4k vcf file. Multi-allelic variants were also removed prior to analysis by VarCount. A diagram showing the steps involved in processing and analysing the variant files is shown in (Figure 1).
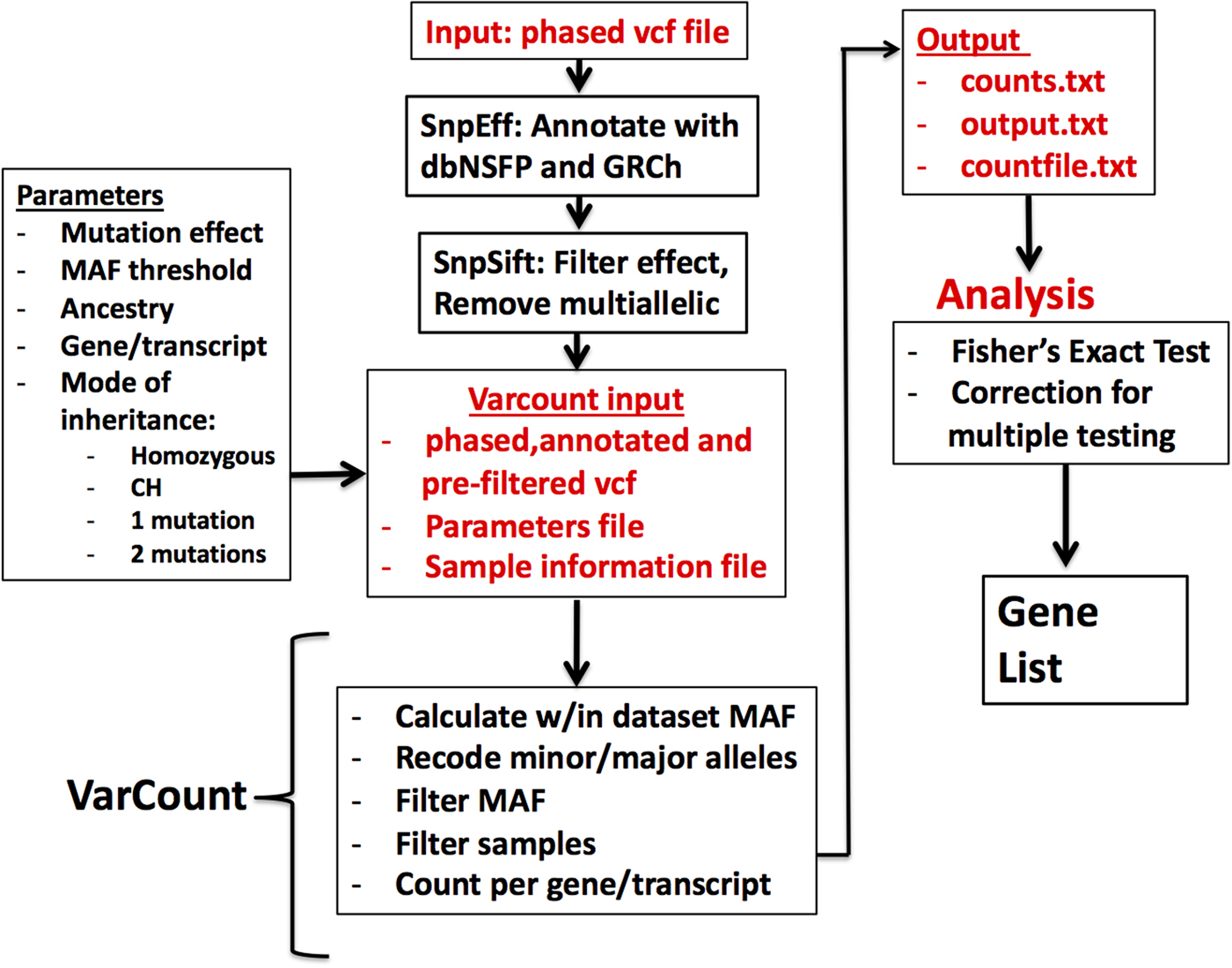
Fig. 1. Flow diagram for the processing and analysis of variant lists. Vcf files are annotated and filtered using SNPSift/SNPEff. Final vcf along with parameter and sample information files are input to VarCount. The input files are processed to recode minor and major alleles when the MAF >0.5 and to count the number of individuals with variants qualifying based on information in the parameter file. The final output lists for every transcript or gene, the number of individuals with qualified variants in that locus (counts.text), which individuals have the variant(s) (countfile.txt), and which variants are harboured by each individual (output.txt).
Vcf files were queried for homozygous and CH variants using VarCount. Because the variants in the 1000 Genomes Project vcf files are phased, determining true compound heterozygotes is automatic using VarCount. In addition to the annotated vcf file, the parameters.txt and subjectinfo.txt (containing sex and ancestry information) were used as input. Within the parameters file, the same qualifications used in the Epi4k analysis were selected: analyses were run for each of the two MAFs (0.5 and 1.0%) and for all 1000 Genomes Project participants and using only those of European (EUR) ancestry. The final output from analyses was for each MAF cutoff and for each population, counts for every transcript in which at least one individual harboured recessive variants.
(iii) Epi4k statistical analysis
Using R statistical software, a Fisher's exact test was used to detect transcripts with significant differences in the proportion of individuals with homozygous or CH variants between the Epi4k dataset and the 1000 Genomes Project dataset. Odds ratios and p-values were calculated using the number of individuals with and without qualifying variants. Analyses were performed using all ancestries, and for only individuals of European ancestry. Both Bonferroni and Benjamini-Hochberg adjustments were used to determine significance thresholds after correction for multiple testing. The number of tests was based on the number of transcripts with at least one individual in either the Epi4k or 1000 Genomes Project dataset with in trans coding variants with MAFs below the set threshold.
(iv) Structural modelling of PRTG
The three-dimensional structure of Protogenin (PRTG) was modelled off the crystal structure of the human receptor protein tyrosine phosphatase sigma (PDB: 4PBX; 25.1% sequence identity) using MODELLER 9.14 (Webb and Sali, Reference Webb and Sali2016). The resultant model superimposed with the template had an RSMD of 4.94 Å over 442 Cα atoms. Charges and hydrogen atoms were added to the wild-type and mutant FGR models using PDB2PQR (Dolinsky et al., Reference Dolinsky, Nielsen, McCammon and Baker2004). Electrostatic potentials were calculated using APBS (Konecny et al., Reference Konecny, Baker and McCammon2012) as described previously (Moshfegh et al., Reference Moshfegh, Velez, Li, Bassuk, Mahajan and Tsang2016; Cox et al., Reference Cox, Darbro, Laxer, Velez, Bing, Finer, Erives, Mahajan, Bassuk and Ferguson2017; Toral et al., Reference Toral, Velez, Boudreault, Schaefer, Xu, Saffra, Bassuk, Tsang and Mahajan2017). Protein and solvent dielectric constants were set to 2.0 and 78.0, respectively. All structural figures were generated by PyMOL (https://pymol.org/2/; Schrödinger, LLC).
3. Results
(i) Varcount: variant quantification at the individual level
Varcount is a free, open source tool useful for the quantification of heterozygous, homozygous or CH variants per sample. Input variants may be phased or unphased. All python scripts and supporting files may be downloaded from Github at https://github.com/GeneSleuth/VarCount. Supporting files include the ‘parameters.txt’ file where the user may select variant filters for variant effect, MAF and inheritance pattern (homozygous, CH, one variant or two variants), and sample filters based on information entered into the ‘SampleInfo file’. Input vcf files must be annotated with SNPSift/SNPEff using the dbNSFP and GRCh37/38 databases. A readme file with instructions is also provided. A flow diagram with the steps involved in processing of data is depicted in Figure 1.
(ii) CH variants in Epi4k probands reveal novel epilepsy genes
We used VarCount to query the Epi4k dataset for rare homozygous and CH variants. The Epi4k data are whole exome data from 264 trios with a child affected by epileptic encephalopathy, either infantile spasms (IS) or Lennox-Gastaut Syndrome (LGS) (Epi4k Consortium et al., Reference Allen, Berkovic, Cossette, Delanty, Dlugos, Eichler, Epstein, Glauser, Goldstein, Han, Heinzen, Hitomi, Howell, Johnson, Kuzniecky, Lowenstein, Lu, Madou, Marson, Mefford, Esmaeeli Nieh, O'Brien, Ottman, Petrovski, Poduri, Ruzzo, Scheffer, Sherr, Yuskaitis, Abou-Khalil, Alldredge, Bautista, Berkovic, Boro, Cascino, Consalvo, Crumrine, Devinsky, Dlugos, Epstein, Fiol, Fountain, French, Friedman, Geller, Glauser, Glynn, Haut, Hayward, Helmers, Joshi, Kanner, Kirsch, Knowlton, Kossoff, Kuperman, Kuzniecky, Lowenstein, McGuire, Motika, Novotny, Ottman, Paolicchi, Parent, Park, Poduri, Scheffer, Shellhaas, Sherr, Shih, Singh, Sirven, Smith, Sullivan, Lin Thio, Venkat, Vining, Von Allmen, Weisenberg, Widdess-Walsh and Winawer2013). Counts were performed using individuals of all ancestries or just those of European ancestry (207/264). Individuals from the 1000 Genomes Project were used as controls. The individual counts and p-values for the analyses are listed in (Supplementary Tables 2–5). Including only rare variants (MAFs below 0.5 and 1.0%) determined enrichment for CH variants in six genes. For combined ancestries, the six genes are in order of significance: OSBP2, PRTG, ABCC11, MACF1, STAB1 and TNC. PRTG and TNC were also highly ranked in the 1% MAF analysis, with one additional count for each transcript. Variants for all six genes are listed in Table 1. In our analysis of just individuals of European ancestry, MACF1 was the most significantly enriched gene using a 0.5% MAF. The p-values indicated in Table 1 are for individual tests; there were no p-values significant after correction for multiple testing.
Table 1. Rare (<0.5 and 1.0% minor allele frequency) compound heterozygous variants in Epi4k participants.
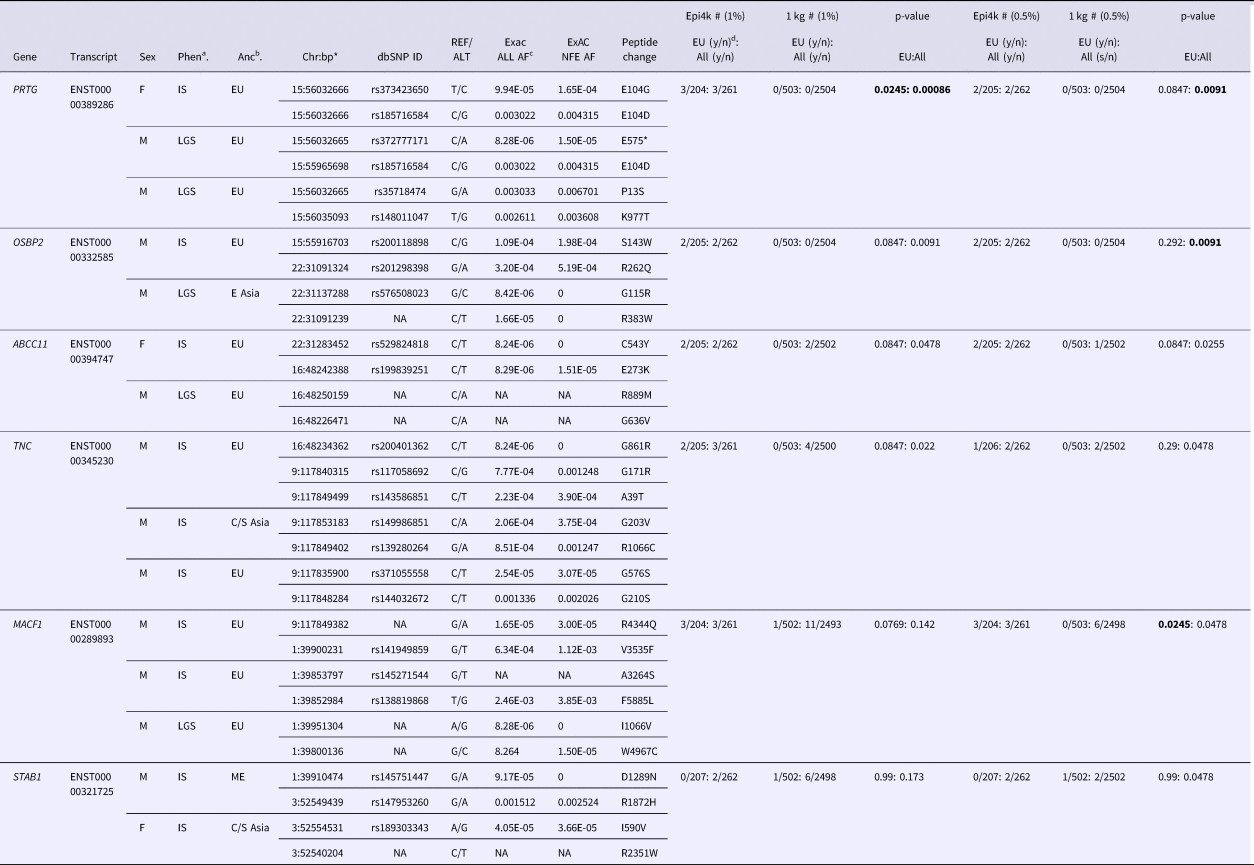
aPhen = phenotype, bAnc = ancestry, cAF = allele frequency, dy/n corresponds to yes/no counts of individuals with qualifying variants.
*bp (base pair position) in hg19/Build37.
Ancestries: EU = European, E Asia = East Asia, C/S Asia = Central/South Asia, ME = Middle East. Phenotypes: IS = Infantile spasms, LGS = Lennox-Gastaut syndrome.
p-values in bold are the most significant for the specific analysis.
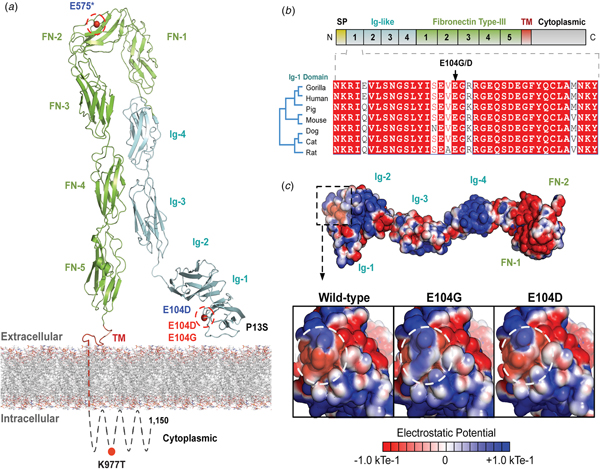
The variants for the three individuals with CH PRTG variants are depicted in Figure 2a. Because of the concentration of variants at position E104, we performed structural modelling to predict the pathogenicity of the PRTG variants. The p.Glu104Gly and p.Glu104Asp variants localize to the immunoglobulin (Ig)-like domain 1 (Figure 2a). Ig-like domains are responsible for mediating protein–protein and protein–peptide interactions. The p.Glu104Gly disrupts a negative charge in the Ig-like 1 domain. This loss of charge may disrupt interactions with putative PRTG-binding partners (Figure 2c).
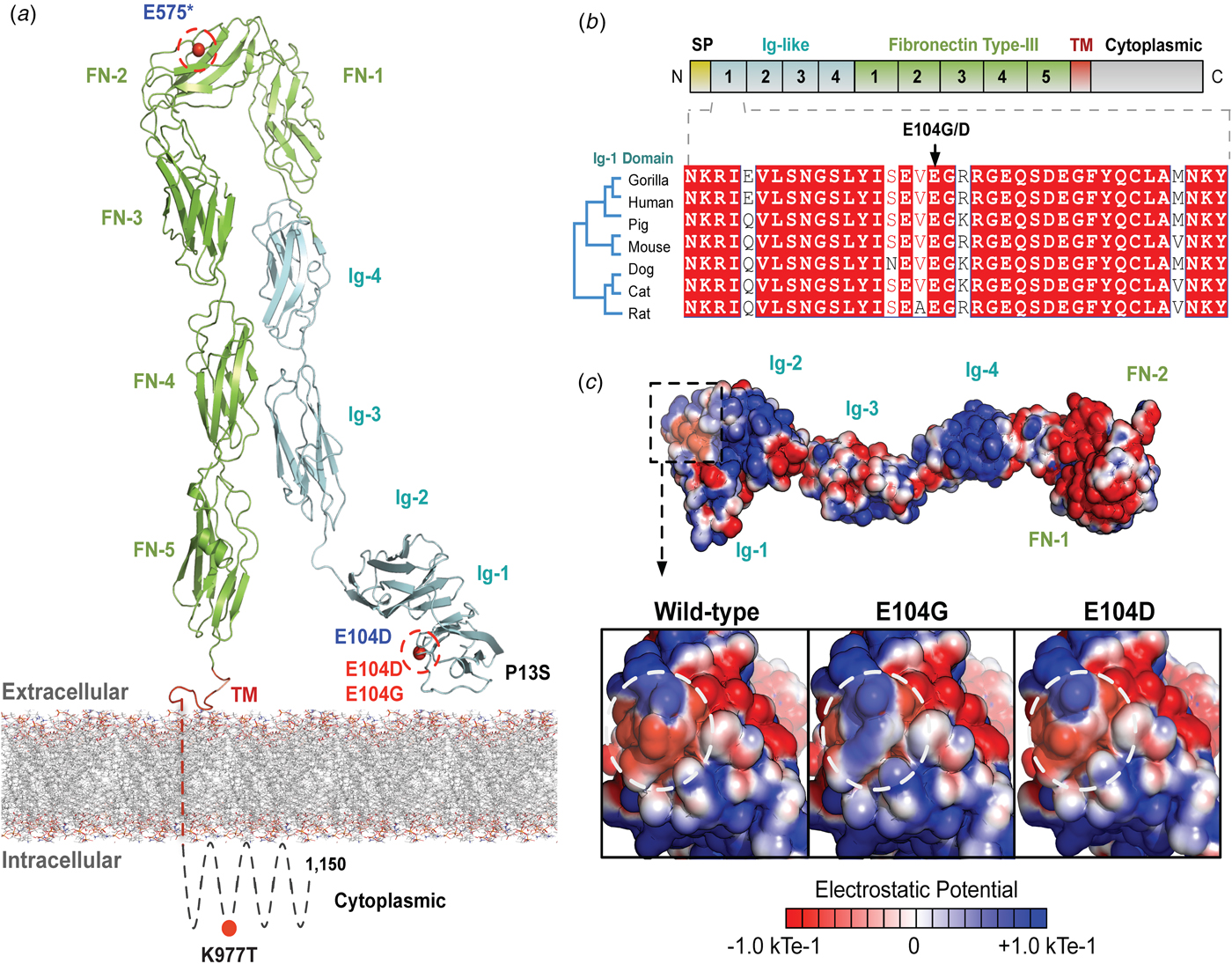
Fig. 2. PRTG compound heterozygous mutations in Epi4k probands. (a) Theoretical model of the human PRTG structure spanning the plasma membrane indicating mutation locations in each child. The three pairs of in trans mutations, indicated in red, were found using a <1% MAF threshold. (b) Schematic representation of PRTG functional domains. Multiple sequence alignment of the PRTG Ig-1 domain. The E104 residue is 100% conserved across seven species. (c) Top: Electrostatic potential surface of PRTG calculated in APBS. Bottom: Close-up of the PRTG electrostatic potential surface at the site of mutation. The p.Glu104Gly mutation leads to a loss of negative charge, which may disrupt interactions with putative PRTG binding partners. The p.Glu104Asp mutation does not lead to a change in charge or electrostatic potential.
The de novo variants identified by Epi4k Consortium and the Epilepsy Phenome/Genome Project (Epi4k Consortium et al., Reference Allen, Berkovic, Cossette, Delanty, Dlugos, Eichler, Epstein, Glauser, Goldstein, Han, Heinzen, Hitomi, Howell, Johnson, Kuzniecky, Lowenstein, Lu, Madou, Marson, Mefford, Esmaeeli Nieh, O'Brien, Ottman, Petrovski, Poduri, Ruzzo, Scheffer, Sherr, Yuskaitis, Abou-Khalil, Alldredge, Bautista, Berkovic, Boro, Cascino, Consalvo, Crumrine, Devinsky, Dlugos, Epstein, Fiol, Fountain, French, Friedman, Geller, Glauser, Glynn, Haut, Hayward, Helmers, Joshi, Kanner, Kirsch, Knowlton, Kossoff, Kuperman, Kuzniecky, Lowenstein, McGuire, Motika, Novotny, Ottman, Paolicchi, Parent, Park, Poduri, Scheffer, Shellhaas, Sherr, Shih, Singh, Sirven, Smith, Sullivan, Lin Thio, Venkat, Vining, Von Allmen, Weisenberg, Widdess-Walsh and Winawer2013) in the nine probands with either PRTG, TNC or MACF1 recessive variants are described in Table 2. For the three patients with CH PRTG variants, one patient harbours a de novo missense variant in HSF2, the second has a nonsense variant in CELSR1, and the third patient has two de novo variants – a missense in Fam102A and a 3´UTR variant in USP42. De novo mutations were only reported in one of the probands with in trans TNC variants – a missense variant in DIP2C and a splice donor change in IFT172. All three patients with CH variants in MACF1 were reported to have de novo mutations. The first patient has a 5´ and 3´UTR de novo variant in FAM19A2 and GLRA2, respectively, and the second patient also has a 3´UTR de novo change in the gene LRRC8D and a missense change in SNX30. One de novo variant was identified in the third proband in the gene FAM227A. Polyphen2 categories and CADD scores for each de novo variant as well as missense and loss-of-function constraint metric values for each gene (from ExAC) are also listed in Table 2. The z-score is a ratio of expected to identified missense variants in a particular gene, and pLI is a gene's probability of being loss-of-function intolerant. These constraint metrics are calculated using genomic data from controls without severe genetic diseases in the ExAC database (Lek et al., Reference Lek, Karczewski, Minikel, Samocha, Banks, Fennell, O'Donnell-Luria, Ware, Hill, Cummings, Tukiainen, Birnbaum, Kosmicki, Duncan, Estrada, Zhao, Zou, Pierce-Hoffman, Cooper, DePristo, Do, Flannick, Fromer, Gauthier, Goldstein, Gupta, Howrigan, Kiezun, Kurki, Moonshine, Natarajan, Orozco, Peloso, Poplin, Rivas, Ruano-Rubio, Ruderfer, Shakir, Stenson, Stevens, Thomas, Tiao, Tusie-Luna, Weisburd, Won, Yu, Altshuler, Ardissino, Boehnke, Danesh, Roberto, Florez, Gabriel, Getz, Hultman, Kathiresan, Laakso, McCarroll, McCarthy, McGovern, McPherson, Neale, Palotie, Purcell, Saleheen, Scharf, Sklar, Patrick, Tuomilehto, Watkins, Wilson, Daly and MacArthur2015).
Table 2. De novo variants in Epi4k probands with compound heterozygous variants in PRTG, TNC or MACF1.
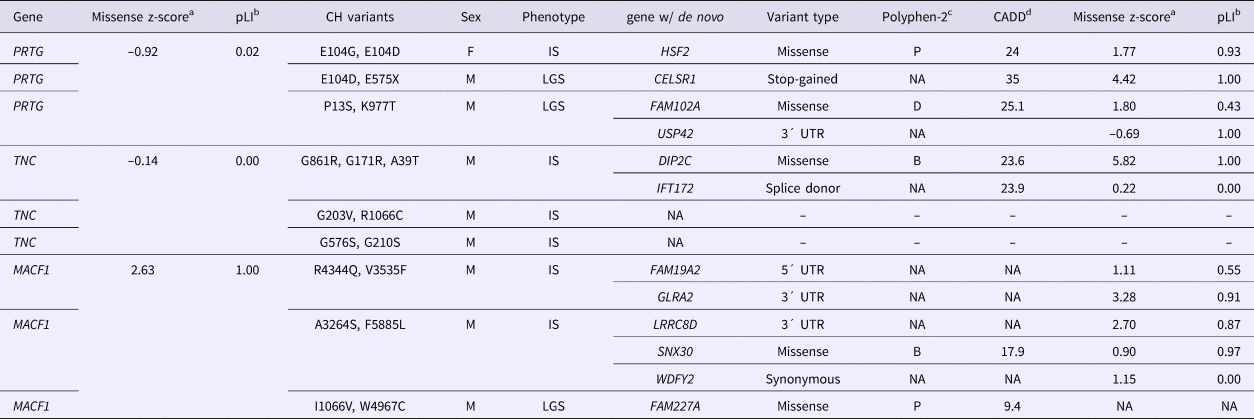
az-score is a measure of tolerance to missense variants, based on ratio of expected to identified; bpLI is the probability that a gene is intolerant to loss-of-function variants; cPolyphen2 – prediction of a missense variant's impact on protein structure and function: B = benign, P = possibly damaging, D = damaging (Adzhubei et al., Reference Adzhubei, Schmidt, Peshkin, Ramensky, Gerasimova, Bork, Kondrashov and Sunyaev2010); dCADD = phred-scaled score of Combined Annotation Dependent Depletion, a measure of the deleteriousness of a SNP or INDEL (Kircher et al., Reference Kircher, Witten, Jain, O'Roak, Cooper and Shendure2014).
Phenotypes: IS = Infantile spasms, LGS = Lennox-Gastaut syndrome.
4. Discussion
Epileptic encephalopathies are a group of severe, early-onset seizure disorders with consistent EEG abnormalities that over time interfere with development and cause cognitive decline (Covanis, Reference Covanis2012). The Epi4k dataset contains exome sequences from 264 trios that include a proband with epileptic encephalopathy, either LGS or IS. LGS is characterized by frequent, mixed epileptic seizures that arise most frequently between the ages of 3 and 5 (Amrutkar & Riel-Romero, Reference Amrutkar and Riel-Romero2018). IS occurs during the first year of life and is cryptic in its presentation, with mild head bobbing and is often not detected until the seizures have caused significant neurological damage (Kossoff, Reference Kossoff2010). IS often progress into LGS over time.
We developed a free and user-friendly tool, VarCount, to query vcf files for individuals harbouring variants that qualify according to user specification. To test its function, we used VarCount to quantify rare, CH variants in probands from the Epi4k trio dataset and found enrichment for variants in six genes including PRTG, TNC and MACF1. PRTG codes for protogenin, a member of the immunoglobulin superfamily that is involved in axis elongation and neuronal growth during early vertebrate development (Toyoda et al., Reference Toyoda, Nakamura and Watanabe2005; Vesque et al., Reference Vesque, Anselme, Couve, Charnay and Schneider-Maunoury2006). TNC and MACF1 are also directly involved in neuronal development and/or growth. TNC (Tenascin-C) is an extracellular matrix glycoprotein involved in axonal growth and guidance (Jakovcevski et al., Reference Jakovcevski, Miljkovic, Schachner and Andjus2013). Seizures up-regulate TNC in the hippocampus, and in a pilocarpine epilepsy model up-regulation was shown to be mediated by TGF-β signalling (Mercado-Gomez et al., Reference Mercado-Gomez, Mercado-Gómez, Landgrave-Gómez, Arriaga-Avila, Nebreda-Corona and Guevara-Guzmán2014). MACF1 is a cytoskeletal crosslinking protein highly expressed in the brain and is crucial for neuron development and migration (Moffat et al., Reference Moffat, Ka, Jung, Smith and Kim2017). MACF1 variants are associated with the neurological pathologies Parkinson's disease, autism and schizophrenia (Moffat et al., Reference Moffat, Ka, Jung, Smith and Kim2017). Recently, highly penetrant de novo MACF1 mutations were identified in several patients with a newly characterized lissencephaly with a complex brain malformation (Dobyns et al., Reference Dobyns, Aldinger, Ishak, Mirzaa, Timms, Grout, Dremmen, Schot, Vandervore, van Slegtenhorst, Wilke, Kasteleijn, Lee, Barry, Chao, Szczałuba, Kobori, Hanson-Kahn, Bernstein, Carr, D'Arco, Miyana, Okazaki, Saito, Sasaki, Das, Wheeler, Bamshad and Nickerson2018). This new phenotype highlights MACF1 variants’ variable impact on disease pathogenesis. Given both the enrichment in Epi4k probands for CH variants in these genes as well as their known involvement in neuronal processes, we suggest that PRTG, TNC and MACF1 are candidate recessive epilepsy genes.
The primary publication reporting analysis of the Epi4k trio dataset was a description of de novo mutations in the probands (Epi4k Consorium et al., Reference Allen, Berkovic, Cossette, Delanty, Dlugos, Eichler, Epstein, Glauser, Goldstein, Han, Heinzen, Hitomi, Howell, Johnson, Kuzniecky, Lowenstein, Lu, Madou, Marson, Mefford, Esmaeeli Nieh, O'Brien, Ottman, Petrovski, Poduri, Ruzzo, Scheffer, Sherr, Yuskaitis, Abou-Khalil, Alldredge, Bautista, Berkovic, Boro, Cascino, Consalvo, Crumrine, Devinsky, Dlugos, Epstein, Fiol, Fountain, French, Friedman, Geller, Glauser, Glynn, Haut, Hayward, Helmers, Joshi, Kanner, Kirsch, Knowlton, Kossoff, Kuperman, Kuzniecky, Lowenstein, McGuire, Motika, Novotny, Ottman, Paolicchi, Parent, Park, Poduri, Scheffer, Shellhaas, Sherr, Shih, Singh, Sirven, Smith, Sullivan, Lin Thio, Venkat, Vining, Von Allmen, Weisenberg, Widdess-Walsh and Winawer2013). An analysis of CH variants was also reported, using a MAF cutoff of 0.15%, which is lower than the cutoff used in the work presented here. In this analysis, the parents were used as internal controls, and CH variants in 351 genes were identified, without genome-wide significance. The authors only listed five of the genes which are known to cause Mendelian disorders that include a seizure phenotype – ASPM, CNTNAP2, GPR98, PCNT and POMGNT1. In our analysis using the 1000 Genomes Project participants as controls, enrichment for CH variants was not detected in any of these genes. Using the number of individuals with in trans variants in a gene (transcript) as an indicator function required at least two probands to have qualifying variants in order to detect single-test significance, with complete absence of qualifying variants in controls. It is clear from the analyses using either internal controls or the 1000 Genomes Project as controls that a larger sample size is required to achieve genome-wide significance.
The de novo variants reported by the Epi4k Consortium and the Epilepsy Phenome/Genome Project (Epi4k Consortium et al., Reference Allen, Berkovic, Cossette, Delanty, Dlugos, Eichler, Epstein, Glauser, Goldstein, Han, Heinzen, Hitomi, Howell, Johnson, Kuzniecky, Lowenstein, Lu, Madou, Marson, Mefford, Esmaeeli Nieh, O'Brien, Ottman, Petrovski, Poduri, Ruzzo, Scheffer, Sherr, Yuskaitis, Abou-Khalil, Alldredge, Bautista, Berkovic, Boro, Cascino, Consalvo, Crumrine, Devinsky, Dlugos, Epstein, Fiol, Fountain, French, Friedman, Geller, Glauser, Glynn, Haut, Hayward, Helmers, Joshi, Kanner, Kirsch, Knowlton, Kossoff, Kuperman, Kuzniecky, Lowenstein, McGuire, Motika, Novotny, Ottman, Paolicchi, Parent, Park, Poduri, Scheffer, Shellhaas, Sherr, Shih, Singh, Sirven, Smith, Sullivan, Lin Thio, Venkat, Vining, Von Allmen, Weisenberg, Widdess-Walsh and Winawer2013) in the nine probands with CH variants in PRTG, TNC or MACF1 are described in Table 2. Of the 12 genes with de novo variants identified in the nine patients, three are implicated in neurological disease. CELSR1 is a planar cell polarity gene in which mutations are known to cause neural tube defects including spina bifida (Robinson et al., Reference Robinson, Escuin, Doudney, Vekemans, Stevenson, Greene, Copp and Stanier2012). De novo deletions of DIP2C have been reported in two patients with cerebral palsy, one of whom also had ADHD, and the other had seizures in infancy (Zarrei et al., Reference Zarrei, Fehlings, Mawjee, Switzer, Thiruvahindrapuram, Walker, Merico, Casallo, Uddin, MacDonald, Gazzellone, Higginbotham, Campbell, deVeber, Frid, Gorter, Hunt, Kawamura, Kim, McCormick, Mesterman, Samdup, Marshall, Stavropoulos, Wintle and Scherer2018). In another report, deletions including DIP2C and/or ZMYND11 were identified in several patients with developmental delay including three patients with seizures (DeScipio et al., Reference DeScipio, Conlin, Rosenfeld, Tepperberg, Pasion, Patel, McDonald, Aradhya, Ho, Goldstein, McGuire, Mulchandani, Medne, Rupps, Serrano, Thorland, Tsai, Hilhorst-Hofstee, Ruivenkamp, Van Esch, Addor, Martinet, Mason, Clark, Spinner and Krantz2012). GLRA2 is a glycine receptor involved in neurodevelopment in which variants are implicated in autism (Pilorge et al., Reference Pilorge, Fassier, Le Corronc, Potey, Bai, De Gois, Delaby, Assouline, Guinchat, Devillard, Delorme, Nygren, Råstam, Meier, Otani, Cheval, James, Topf, Dear, Gillberg, Leboyer, Giros, Gautron, Hazan, Harvey, Legendre and Betancur2016; Lin et al., Reference Lin, Xiong, Li, Gong, Cao, Kuang, Zhang, Gao, Mechawar, Liu and Zhu2017), including a patient with comorbid epilepsy (Zhang et al., Reference Zhang, Ho, Harvey, Lynch and Keramidas2017).
Of the de novo variants reported in these genes, the nonsense variant in CELSR1 identified in one of the probands with in trans PRTG variants is the most likely to be pathogenic. However, regarding their involvement in neural tube defects, variants in CELSR1 are thought to contribute to pathogenesis but not in a Mendelian fashion, as variants have been found to be inherited from unaffected parents or to be ineffective in functional assays (Robinson et al., Reference Robinson, Escuin, Doudney, Vekemans, Stevenson, Greene, Copp and Stanier2012; Allache et al., Reference Allache, De Marco, Merello, Capra and Kibar2012). The nonsense CELSR1 variant in the patient reported here may contribute to epilepsy in the presence of a genetic modifier. The de novo missense mutation in DIP2C is predicted to be deleterious (CADD = 23.6) and has a low rate of benign missense variation based on constraint metrics (z = 5.82). The de novo variant in GLRA2 is in the 3´UTR so it is difficult to predict its impact on gene function and subsequent pathogenicity.
The CH variants in PRTG, TNC and MACF1 are similarly variable in predicted pathogenicity, with CADD scores ranging from between less than one to 38. PRTG and TNC both have constraint metrics indicative of a high tolerance to both missense and loss-of-function variants, while MACF1 is moderately intolerant of missense variants (z = 2.63) and extremely intolerant of loss-of-function variants (pLI = 1.0). Interestingly, aside from the 3´UTR variant in GLRA2, none of the de novo variants in the Epi4k participants with MACF1 CH variants are in genes associated with neurological disease or predicted with confidence to have a negative impact on gene function. This, in addition to MACF1’s intolerance to missense or nonsense variants, is supportive of the pathogenicity of the biallelic variants in the gene.
In summary, we present a free tool VarCount for the quantification of qualifying variants as an indicator function per individual in the analysis of variant lists (vcf files). We used VarCount to assess enrichment of rare, coding, CH variants in a cohort of 264 epilepsy probands and found enrichment in three genes involved in neurodevelopmental processes – PRTG, TNC and MACF1. A missense change at the E104 residue of PRTG was identified three times in two different probands. Significance was not maintained after correction for multiple testing, and larger cohorts or candidate gene studies using a different sample set are necessary to validate this enrichment. In the context of the de novo mutations also present in these patients, experimentation is necessary in order to delineate if the CH or de novo variants, or both, are pathogenic in the development of epileptic encephalopathy. PRTG, TNC and MACF1 are candidate recessive epilepsy genes and our work highlights that inheritance of CH variants should not be excluded from gene discovery or diagnostic analyses of patients with epilepsy.
Author ORCIDs
Allison J. Cox, https://orcid.org/0000-0002-6803-4456
Acknowledgements
This work was supported by the following grants: T32GM008629 (AJC), T32GM082729-01 (AJC), T32GM007337 (FG and GV), R01AR059703 (PJF, VBM and AGB) and R01NS098590 (AGB, PJF).
Declaration of interest
None.
Supplementary material
For supplementary material accompanying this paper visit http://dx.doi.org/10.1017/S0016672319000065






 Open access
Open access