


1. Introduction
It has been known since Robbins (Reference Robbins1918) that linked genes, even closely linked ones, are expected to be associated at random (linkage equilibrium) in an infinite population. Selection for particular gene combinations may lead to linkage disequilibrium (LD) (see Lewontin & Kojima, Reference Lewontin and Kojima1960; Franklin & Lewontin, Reference Franklin and Lewontin1970), but such LD is only important if selective interactions are widespread.
Hill & Robertson (Reference Hill and Robertson1968), Sved (Reference Sved1968) and Ohta & Kimura (Reference Ohta and Kimura1969) first drew attention to the importance of the infinite population size assumption and showed that closely linked genes necessarily become strongly associated because of genetic drift in a small population. Prior to this, Haldane (Reference Haldane1949) had pointed out the conceptually similar result that inbreeding leads to the association of genotypes at linked loci, a result extended by Bennett & Binet (Reference Bennett and Binet1956) and other authors for the inbreeding system of mixed self-fertilization and random mating.
Arguments that LD is not to be expected are now of historical interest only. Following the cloning and sequencing of many gene regions, it has become clear that LD of closely linked nucleotide sites is the norm (see e.g. Conrad et al., Reference Conrad, Jakobsson, Coop, Wen, Wall, Rosenberg and Pritchard2006). This is evidently expected in cases of disease-causing genes in humans where all mutations in a population trace back to a single source and provides the basis for LD mapping (see e.g. de la Chapelle & Wright, Reference de la Chapelle and Wright1998).
The widespread occurrence of LD has made it of interest to consider the general problem of the expectation for the amount of LD as determined by the balance between genetic drift and recombination in a population. A number of authors have given expectations, e.g. Hill & Robertson, Reference Hill and Robertson1968; Sved, Reference Sved1968, Reference Sved1971; Ohta & Kimura, Reference Ohta and Kimura1969; Serant & Villard, Reference Serant and Villard1972; Littler, Reference Littler1973; Weir & Cockerham, Reference Weir and Cockerham1974; Hill, Reference Hill1977; Vitalis & Couvet, Reference Vitalis and Couvet2001a, Reference Vitalis and Couvetb. These expectations are considered further in the following section.
The purpose of the present paper is to extend the LD analysis to the case of population subdivision. The analysis uses as a measure of LD the correlation of gene frequencies, r (Hill & Robertson, Reference Hill and Robertson1968). Within populations, LD is measured as normally using the parameter r 2. As a convenient measure of the association of LD between populations, e.g. for populations i and j, the parameter rirj is introduced. Note that r 2 and rirj are, respectively, the variance and covariance of r values over replicate populations, given that E(r)=0. It is shown that the combination of the within- and between-population measures allows a straightforward analysis in terms of migration, recombination and population size.
The formulae derived in the present paper are applied to the case of current human populations. The model of interest is one in which population subdivision occurs at a particular point in time, and the subdivision persists for periods of time that are small compared with evolutionary time units. Mutation comes into the model only to the extent that initial gene frequencies and LD levels at the time of population subdivision may be a product of past mutation rates and population size. Following population subdivision, it is assumed that the effects of mutation can be ignored compared with the effects of genetic drift, recombination and migration. Hapmap data examining the divergence between African and non-African populations are used to illustrate the methods.
2. Analysis
(i) The model for a single population
Loci A and B are assumed to be linked with recombination frequency c in a population of size N e reproducing according to the Wright–Fisher model. The initial calculation is in terms of identity-by-descent probabilities. This is then related to frequencies, specifically to the LD measure r.
The concept of linked identity-by-descent (LIBD) was introduced in Sved (Reference Sved1971). LIBD refers to the event in which genes at linked loci of two haplotypes in a population are identical-by-descent through the same pathways, i.e. without recombination, from an ancestral haplotype (see Fig. 1). The probability of LIBD will be denoted as L, replacing Q used in Sved (Reference Sved1971), which was defined using a conditional probability of LIBD in a bi-allelic population. Note that L differs from two locus identity parameters such as X 11 (Weir & Cockerham, Reference Weir and Cockerham1974), θ (Tachida & Cockerham, Reference Tachida and Cockerham1986), F 11, (Goodnight, Reference Goodnight1987; Whitlock et al., Reference Whitlock, Phillips and Wade1993) and φ (Vitalis & Couvet, Reference Vitalis and Couvet2001a), all of which denote IBD at two loci irrespective of the pathways by which this is attained. LIBD implies a much-restricted subset of such pathways.

Fig. 1. Pathways for identity-by-descent for one and two loci.
The recurrence relationship for L between generations can readily be written down. The model is formally analogous to the case of a single locus, with recombination in the two-locus model replacing mutation in the single locus model. The LIBD probability in the offspring generation in terms of L in the parent generation is (Sved & Feldman, Reference Sved and Feldman1973).
The equation is readily generalized to any number of generations. The value of L at equilibrium comes to 1/[1+(2N e−1)(2c−c 2)], which is approximately 1/[1+4N ec].
L is a parameter in terms of probabilities. The usual measures of LD such as D or r are frequency parameters. Sved (Reference Sved1971) used a roundabout argument to show that the expected value of r 2 is equal to L. A related but simpler argument was given by Sved & Feldman (Reference Sved and Feldman1973), using an analogy with single locus inbreeding arguments as shown here.
The single locus argument can be seen using the pathways of Fig. 1, focusing on the A locus (I). The coefficient of inbreeding was initially defined by Sewall Wright (see e.g. Wright, Reference Wright1931) in terms of the correlation between uniting gametes. Somewhat later, the identity-by-descent probability definition of inbreeding was adopted by Malécot (Reference Malécot1948). The relationship between the correlation and probability definitions may be seen in the following simple way, closely related to the argument of Crow & Kimura (Reference Crow and Kimura1970, p. 66). If the genes are identical by descent, then the correlation is 1. If they are not identical by descent, then the correlation is 0. In terms of the probability of IBD, fA, the overall correlation rA then becomes fA1+(1−fA)0=fA. This implies that correlations are additive, an assumption that can be checked by writing out the full set of matings (Crow & Kimura, Reference Crow and Kimura1970, Table 3.2.1).
The equivalent two-locus argument can be seen by following the pathways labelled (II) in Fig. 1. The probability that the A and B alleles are transmitted intact (without recombination) on either pathway from the common ancestral gamete can be defined as fAB. In such an event, the correlation is equal to 1. Any recombination event is assumed to connect the A allele to a random B allele in the population, assuming random mating, so that the correlation will be 0. The overall correlation is equal to
The LIBD probability L defined previously is equal to ![]() , since events in the two pathways leading to the present gametes are independent. So the relationship between r and L can be given as
, since events in the two pathways leading to the present gametes are independent. So the relationship between r and L can be given as
where the expectation is over replicate populations with the same probability structure.
This allows us to write the expectation for r 2 in the offspring generation in terms of r 2 in the parent generation. Using eqn (1).
The exact validity of eqn (2) is in some doubt following arguments by Littler (Reference Littler1973) and McVean (Reference McVean2002) questioning whether LIBD arguments involving just the current population can account for the historical complexity. However, computer simulation shows that the equation holds with reasonable accuracy provided N e is not too small. Further support for eqn (2) comes from an alternative derivation by Tenesa et al. (Reference Tenesa, Navarro, Hayes, Duffy, Clarke, Goddard and Visscher2007), based on properties of the correlation coefficient rather than LIBD arguments. Hill & Robertson (Reference Hill and Robertson1968) also derived the expected equilibrium value of r 2 as 1/4N ec, based on a balance between gain of LD by drift and loss by recombination. Hill (personal communication) has pointed out that a simple modification of this derivation yields the same equilibrium result as for eqn (2), E[r 2]=1/(1+4N ec).
(ii) Population subdivision
For a single closed population, the parameter L denotes the probability that two gametes sampled, with replacement, from the population are identical through the same pathways, i.e. without recombination. For an island model of population subdivision, L W becomes the LIBD probability for two gametes sampled from the same island population, while L B is the LIBD probability for two gametes sampled from different islands.
Sampling with replacement within a population is assumed in the models. This may seem a somewhat artificial construct compared with the manner in which identity-by-descent is traditionally defined in terms of sampling of two different uniting gametes (Wright, Reference Wright1931). However, sampling with replacement is formally consistent with the equation of probability and frequency parameters when quantities such as p 2, r 2, etc., are considered (Sved & Latter, Reference Sved and Latter1977). In practice, the differences between sampling with and without replacement are small.
The usual parameters for the island model of population subdivision are assumed. The overall population is divided into k islands, each of which exchanges genes at an equal rate with each other island. The overall rate of immigrant genes into any island population is m per generation, randomly sampled from other islands. The parameters N e and c describe the effective population size per island and the recombination frequency, respectively.
Recurrence relationships can be given for the quantities L W and L B in terms of the equivalent parameters of the previous generation. The exact relationship depends on the gene exchange and census model adopted. For example, it is possible to assume deterministic gene exchange, exchange of a fixed number of genes or individuals or exchange of variable numbers of genes or individuals (Latter & Sved, Reference Latter and Sved1981). Furthermore, the population may be censused before or after gene exchange.
This treatment assumes a model in which stochastic sampling according to the Wright–Fisher model occurs within each island population, followed by the exchange of a variable number, but with fixed probability, of migrant genes between islands. This is the same as assuming a deterministic exchange of genes between islands, followed by stochastic sampling (Sved & Latter, Reference Sved and Latter1977). Census of the population is assumed to take place following sampling and gene exchange.
(iii) LIBD probabilities for a subdivided population
The recurrence relations for the island model may be written as follows (cf. Maruyama (Reference Maruyama1970), Latter (Reference Latter1973) and Latter & Sved (Reference Latter and Sved1981) for the case of a single locus):

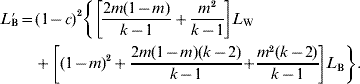
The justification of (3) is as follows. The constant term in (3), 1/2N e, represents the probability that the same gamete is sampled twice. Recombination does not enter into the probability of LIBD for this term. The term L W in (3) involves a contribution from cases in which both gametes sampled are non-immigrant, (1−m)2, added to the probability of both gametes being immigrant but independently from the same island, m 2/(k−1). Both of these terms must be multiplied by the term (1−c)2, representing the probability of non-recombination in both gametes. The contribution from the L B term in (3) consists of the complementary cases in which the two gametes come from different islands, multiplied by the same recombination term. The justification of (4) is similar. In this case, the same gamete cannot be sampled twice, and the only distinction is between gametes that were in different islands before migration, for which the appropriate multiplier is L B, or in the same island, in which case the multiplier is L W.
Equations (3) and (4) may be rewritten as
where
and
Equilibrium solutions may be obtained by setting ![]() and
and ![]() . On the assumption that the quantities m and c are small compared with 1, we may make the approximations:
. On the assumption that the quantities m and c are small compared with 1, we may make the approximations:
All equations that follow are therefore approximations from this point of view. Equations (5) and (6) lead to the equilibrium solutions
and
Equations (8) and (9) can be expressed in the alternative form:
and
where ρ=m/m+(k−1)c is a convenient measure of the ratio of gene flow to recombination.
Equations (10) and (11) have the expected properties at the extremes of gene flow. When there is no gene flow, m=0 and ρ=0, the island model should reduce to the single population model. As expected, eqn (10) reduces to
Similarly there should be no identity between islands, and ![]() is equal to zero, as expected.
is equal to zero, as expected.
When gene flow is large compared with recombination, ρ=1, and eqn (10) reduces to
This is as expected in an overall random mating population of the same effective size as the combined islands (kN e). Also, the identity of gametes from different islands is similar to the identity within islands in this case.
(iv) Predictions of LD
The key argument of Fig. 1 is that correlations equate directly to probabilities of descent without recombination. The expected value of r 2 within populations is equal to the product of probabilities of descent without recombination of two gametes chosen from the same population, or L W. Similarly the expected product of correlations between populations is equal to the product of probabilities of descent without recombination of two gametes chosen from different populations, or L B, Thus
and
where i and j are different islands.
Substituting from eqns (10) and (11) gives the equilibrium solutions as
and
These are more accurately described as ‘steady state’ rather than equilibrium solutions, since fixation at either or both loci will eventually occur in the absence of mutation, in which case correlation values are indeterminate.
The values of E(r 2) and E(rirj) at any time during the process may be found by substitution into eqns (5) and (6), giving the recurrence relationships:

where α and β are as defined in terms of m and k in conjunction with eqns (5) and (6).
3. Computer simulation
(i) Assumptions
The variety of possible parameters in the island model, N, k, m, c, in addition to allele frequencies and LD levels, makes it difficult to completely test the predictions of the above formulae. An initial decision was made to restrict the simulations to the case where all island populations started with the same frequencies. Such an assumption would, for example, appear to be appropriate for studying the divergence of current human populations. An alternative model is one in which allele frequencies in individual island populations are determined by the production of new alleles by mutation and the loss by drift. Such a model, however, requires the existence of stable island populations over evolutionary time periods. In the analysis of currently subdivided populations it seems more realistic to postulate a model in which the island populations are formed by subdivision of an ancestral population in which all alleles already exist at the start of the simulation.
The simulations reported below considered only two island populations. The addition of more populations, up to 16, did not change any conclusions.
Forward computer simulation to test recurrence formulae requires that initial allele frequencies and levels of LD be specified. A range of possibilities was studied by using initial allele frequencies as either 0·5 or 0·05 at each locus. For each combination of allele frequencies, populations were set up with extremes of LD, either in linkage equilibrium (r 2=0) or with the highest value of r 2 consistent with the allele frequencies. In reality, expectations over many locus pairs are expected to involve summation over a range of allele frequencies and levels of LD. This situation was modelled using starting populations from a steady state infinite site mutation model with the same values of N and c as assumed in the subdivision simulation.
A range of m values was used, from Nm=1/4 to Nm=64. Generations were simulated using a Wright–Fisher model. Runs were carried out until either fixation occurred at either locus or a steady state was reached. Replication over a large number of simulations showed that a steady state of r 2 and r 1r 2 values was reached after 3–4000 generations.
The computer simulation was implemented in two different ways. In the first, haplotype frequencies for each island population were calculated deterministically using haplotype frequencies of the previous generation and the relevant recombination and gene flow parameters. Gametes were then sampled at random up to the requisite population number using the calculated frequencies. The second simulation was entirely stochastic, with gamete choice, recombination and migration each occurring stochastically. As expected (Sved & Latter, Reference Sved and Latter1977), the two implementations generated very similar results.
(ii) Results
Simulations starting from different allele frequencies and either zero or maximum levels of LD generated a range of outcomes. In general, simulations starting from central allele frequencies (0·5) showed good agreement between observed and expected values, while non-central frequencies (0·05) gave worse agreement (results not shown). Because of the variety of outcomes, it seemed more informative to use the simulations starting from a range of frequencies and LD values given by the infinite site mutation model.
The simulations presented below were carried out using a value for N of 8192 and c of 1/1024 (Nc=8). The value of N was chosen as consistent with ancestral human population sizes (Tenesa et al., Reference Tenesa, Navarro, Hayes, Duffy, Clarke, Goddard and Visscher2007). The value of c, approximately 0·1 cM, consistent with around 100 kb, is low but sufficiently high to minimize complications of ‘fixation bias’ (Sved et al., Reference Sved, McRae and Visscher2008).
As mentioned above, the simulations use a mixed population of starting allele frequencies. As expected, the results are dependent on the choice of allele frequencies, specifically on the choice of minor allele frequency (MAF). Two levels were chosen for the initial frequencies, 0·01 and 0·3. The latter, based on much older mutations, gave considerably higher starting values of r 2, as well as much lower levels of fixation.
Figure 2 shows the value of r 2 from the simulations together with the expected value from eqns (16) and (17). Values are presented for two levels of migration, low (Nm=1/4) and high (Nm=64), together with the low- and high-MAF values.

Fig. 2. Test for predicted levels of LD within populations for low and high levels of migration and low- and high-MAF values. The predicted (expected) values are shown as either unbroken or broken lines, while the results from the simulations are shown as square or round symbols. Low migration values are shown with broken lines (expected) and unfilled symbols (observed), while high-migration values are shown with unbroken lines and filled symbols. High-MAF values are shown with thicker lines and larger symbols than low-MAF values. Allele frequencies and initial LD levels are from simulations of a single population infinite site mutation model with the same values of N (8192) and Nc (8) assumed in the subpopulation simulation.
Figure 2 shows that the starting point for the simulations is determined by the MAF value. On the other hand, the steady state values of r 2, both observed and expected, are determined essentially by the migration rates. The absolute difference between the steady state values for the two migration rates is not high. The simulation involved only two populations, so that the island population size and the overall population size differed by only a factor of two. When more populations were simulated, the differences between high- and low-migration values were correspondingly larger (results not shown). Note also that the observed and expected values for the two MAF starting points coalesce at much the same time for very different migration rates.
The agreement between observed and expected values is very good for both migration rates for the MAF=0·3 case, and less so for the MAF=0·01 case. It is probably significant that very little fixation occurs in the former case, whereas in the latter around 50% of populations are fixed by the end of the simulation. However, even in the latter case it appears that minor disagreements between the LIBD-derived formulae and simulation results precede any fixation, showing that fixation is not the only reason why the formulae are approximate.
Figure 3 shows the equivalent results for r 1r 2 for the same simulations as in Fig. 2. Note the reversal of the high and low migration outcomes. High migration rates lead to a lower value of r 2, since the overall rather than the local population size becomes the determiner of LD – eqn (12) versus eqn (13). On the other hand, high migration leads to more similarity in r values of different populations, in other words a higher value of r 1r 2. The agreement of simulated values with the expectation given by eqns (16) and (17) is slightly better than for the r 2 values of Fig. 2.

Fig. 3. Values of r 1r 2 measuring the relationship of LD between populations. Symbols and conditions of the simulation are as in Fig. 2.
4. Application to human populations
In the absence of migration, eqn (17) reduces to
This equation is readily generalized to any number of generations (see also de Roos et al., Reference de Roos, Hayes, Spelman and Goddard2008). If a population is subdivided into populations 1 and 2, with initial LD given by r 0, then after T generations, the expected product of r values is
from which the estimate of separation time is obtained as
Sved et al. (Reference Sved, McRae and Visscher2008) used this equation, derived independently using infinite-size population theory, to estimate the age of separation of African and non-African populations. Data came from the Hapmap study (http://www.hapmap.org), and assumed that current African values could be used to estimate ![]() . Locus pairs were pooled in classes based on their estimated recombination frequency. Pooled classes of rirj and r 2 values were used to estimate values of T for the separation of African (YRI) and European (CEU) populations. A separate estimate was obtained for each recombination class ranging from 0·1 to 0·3 cM (filled squares, Fig. 4). Recombination classes below c=0·1 cM are omitted from Fig. 4 because of discrepancies due to ‘fixation bias’ (Sved et al., Reference Sved, McRae and Visscher2008).
. Locus pairs were pooled in classes based on their estimated recombination frequency. Pooled classes of rirj and r 2 values were used to estimate values of T for the separation of African (YRI) and European (CEU) populations. A separate estimate was obtained for each recombination class ranging from 0·1 to 0·3 cM (filled squares, Fig. 4). Recombination classes below c=0·1 cM are omitted from Fig. 4 because of discrepancies due to ‘fixation bias’ (Sved et al., Reference Sved, McRae and Visscher2008).

Fig. 4. Estimated separation time for Europe (CEU) from Africa (YRI). Filled squares show the calculated separation time in generations from applying eqn (19), derived under the assumption of no migration, to the Hapmap data. The straight line shows the values of T obtained from the same equation if actual separation occurred 3000 generations ago and migration occurred throughout at rate m=0·0016. The dashed line shows the values of T obtained from the same equation if actual separation time was again 3000 generations but migration occurred at rate m=0·0128 just for the first 2500 generations.
The striking feature of Fig. 4 is the low estimate of T for all recombination classes, estimates ranging between 600 and 800 generations. Assuming a generation length of 25 years, the separation time translates to between 15 000 and 20 000 years. Current estimates of the time of the most recent migration out of Africa are around 60 000 years (e.g. Cavalli & Feldman, Reference Cavalli-Sforza and Feldman2003).
To explain the discrepancy between the estimates of 20 000 and 60 000 years, Sved et al. (Reference Sved, McRae and Visscher2008) suggested that a reasonably small amount of migration (gene exchange) between populations could severely reduce the estimated divergence time based on LD under the model of no migration. This explanation is now examined in some detail. It is convenient to summarize the results using T estimates obtained from eqn (19). The parameter T is not a true time estimate if migration is included in the model, although it might be thought of as an estimate of ‘effective divergence time’.
Equations (16) and (17) are needed to calculate the expected value of r 1r 2. Equation (16), however, predicts only the expected value of r 2. As mentioned above, in estimating the time of divergence of non-African and African populations (Sved et al., Reference Sved, McRae and Visscher2008), it was assumed that the current value of r 2 in Africa could be used as a predictor of the value of r 2 at the time of population divergence, i.e. that there had been no change over the time of divergence. The validity of this assumption was justified by calculating the expected value of r 2 for various possible values of N e for the African population, showing that they had a minor impact on the estimates of T.
An equivalent assumption has been used in the present calculation. For two populations, with a fixed value of r 02 in population 1 (Africa), eqn (17) becomes
The parameter m refers specifically to migration from population 1 (Africa) to population 2 (Europe), since migration in the opposite direction is ignored. The value of r 22 is dependent on the size of population 2, but does not affect eqn (20).
Equations (19) and (20) were used to calculate values of T for different values of m, over the range c=0·1–0·3 cM under the supposition of a true number of 3000 generations since separation. Values calculated for m=0·00015 gave a mean T close to the calculated mean for Hapmap values. However, as shown in Fig. 4, the slope of estimated T values was much higher than the slope of the observed values. This is a consequence of the fact that the denominator of eqn (19) contains c. Evidently the change in the numerator of eqn (19) is not sufficient to compensate for the increase in the value of c in the denominator over the range 0·1–0·3 cM.
Various models were then examined in which migration was applied over less than the full number of generations. Applying migration only in the latter stages of the divergence increased the slope of the relationship. The most extreme model of this kind is one in which all migration occurs in the final generation, just before measurements are made. It can be shown that in the extreme case where sufficient time has passed so that the expected value of r 1r 2 is zero, migration between the two populations at rate m induces a non-zero correlation in r values, such that the estimated value of T is approximately equal to −ln(2m)/2c. Of course T ceases to be a time estimate at all in such a case.
By contrast, if migration occurs only in the earlier part of the divergence, then the slope of T estimates decreases. Figure 4 shows that migration at the higher rate of m=0·00071 for the first 2650 of 3000 generations, followed by 350 generations with no migration, provides a satisfactory fit to the data.
A common feature of models that explain the data is that migration occurs earlier in the divergence rather than later. Note that any final period with no migration needs to be less than 1000 generations to generate T estimates less than 1000 generations, no matter what the migration rate preceding this period. The figure of 350 generations without migration in Fig. 4 is chosen to provide approximately the correct slope in Fig. 4. However, the assumption of a constant migration rate for the first period, followed by zero for the second period, is obviously only one of a spectrum of models that could provide a fit to the data.
5. Discussion
Values of r 2 and r 1r 2 from the computer simulations agreed reasonably well with expectations when the populations were founded with alleles at central frequencies at both loci. The agreement was worse in the case of populations founded with one or both loci having one allele at low frequency. The simulation with a mixture of frequencies generated by a mutation model constrained to a high MAF value before separation, MAF=0·3, showed levels of agreement not very different from the central frequency simulation.
Fixation of one or more alleles cannot be taken into account by the current LIBD method. The LIBD probability does not take allele frequencies into account, and includes fixed and unfixed populations. By contrast, estimates of LD using r or r 2 cannot take into account populations where fixation has occurred. Furthermore there are obvious biases in the fixation process. Since LD is produced by fluctuation of allele frequencies, populations where such fluctuations are more extreme, and therefore where fixation may tend to occur earlier, are also those populations where high levels of LD are expected. A different type of ‘fixation bias’ has also been discussed by Sved et al. (Reference Sved, McRae and Visscher2008). These effects may be responsible for the discrepancy in the build-up of LD from r 2=0 seen for the low MAF simulations in Fig. 2. As remarked earlier, however, fixation cannot account for all of the discrepancies.
Selection for particular genes combinations would be expected to have a high effect on the migration estimates. Any selective force favouring particular allele combinations will tend to affect values of r in different islands in the same manner. Values of rirj will thus be inflated, and similarly estimates of m if this effect is not recognized.
One other factor that needs to be considered is the possibility of heterogeneous samples. The calculations of the present paper have assumed a simple structure of islands within which mating is at random. If the structure of a subdivided population is less well defined, it may not be possible to recognize within- and between-island contributions. Nei & Li (Reference Nei and Li1973) and Feldman & Christiansen (Reference Feldman and Christiansen1974) have pointed out that if a sample contains contributions from heterogeneous sources then some LD will be found, regardless of whether it exists within the random mating regions. Such LD will not persist over generations except to the extent that exactly the same regions are combined in successive samples. Such sample heterogeneity should also be detectable at the individual locus level through departure from Hardy–Weinberg expectations.
The variation in values between different samples also needs to be emphasized. The observed values of Figs 2 and 3 are based on averages of many thousands of replicate simulations. Any estimated values of migration and effective population size parameters from a single set of populations may thus have extremely high standard errors.
(i) Comparison with previous studies
Three previous studies have looked at LD within subdivided populations (Ohta, Reference Ohta1982a, Reference Ohtab; Tachida & Cockerham, Reference Tachida and Cockerham1986; Vitalis & Couvet, Reference Vitalis and Couvet2001a, Reference Vitalis and Couvetb). Ohta introduced a range of within- and between-population LD parameters by analogy with the IS, IT, ST notation introduced by Wright (Reference Wright1931) to measure inbreeding in a hierarchical manner. Five parameters were introduced, including D IS2 measuring LD within populations and D IT2, D ST2, D ST′2 and D ST′2, measuring various levels of departure of within-population haplotype and gene frequencies compared with overall haplotype and gene frequencies.
Tachida & Cockerham (Reference Tachida and Cockerham1986) introduced a more systematic parameter set. They considered genes on the same gamete, genes on different gametes within an individual, genes on different individuals within a deme, and genes on different demes within the same population. Derivations assumed the Wright–Fisher model in which there is no distinction between genes in the same individual and genes in different individuals within the same deme. As in Ohta (Reference Ohta1982a, Reference Ohtab), expectations were derived for the equilibrium case in a model including mutation, migration and recombination.
The formulation of the present paper differs from that of Ohta (Reference Ohta1982a, Reference Ohtab) and of Tachida & Cockerham (Reference Tachida and Cockerham1986) in the types of population and mutation models. The latter studies are relevant to long-term population descriptions. Ohta considered a model for the past evolution of humans, assuming a population structure of 200 subpopulations of size 100, with substantial migration levels and replacement of subpopulations following extinction. The model assumed a high mutation rate to new alleles. By contrast, the present study is oriented towards migration between current human populations. It considers the level of LD within and between populations starting with subdivision at arbitrary levels of LD, over time intervals sufficiently limited that mutation will not play a role. Although the equations have simple steady state solutions, their applicability is limited in the case where time periods are long enough for mutation to be of importance.
The between-population measure of the present study, rirj, also differs from the between-population measures of previous studies. Although it uses correlation r values rather than the D values of these studies, the corresponding Di Dj statistic does not appear explicitly in their measures. In a model with large numbers of populations, however, the expectation of Di Dj is approximately equal to the demic LD measure of Tachida & Cockerham (Reference Tachida and Cockerham1986).
The model introduced by Vitalis & Couvet (Reference Vitalis and Couvet2001a, Reference Vitalis and Couvetb) is similar to the model of Tachida & Cockerham (Reference Tachida and Cockerham1986), except that a distinction is made between genes in the same individual and genes in different individuals, thereby allowing different degrees of self-fertilization to be taken into account. Explicit measures for between-population LD are not given in this formulation, since their expectation would require 28 parameters (Vitalis & Couvet, Reference Vitalis and Couvet2001a). These authors consider instead optimal procedures for estimating N e and m, assuming a model with an infinitely large number of sub-populations. The extra information available from the combination of single locus and two-locus parameters allows a more accurate estimate of N em compared with that given by the application of eqns (14) and (15), which consider only two-locus LD measures.
(ii) Application to human populations
The recurrence equations have been applied to estimate the time of divergence of human populations (Fig. 4), using data from African (YRI) and non-African (CEU) Hapmap populations. A primary conclusion from this analysis is that the divergence between populations is difficult to explain without invoking some gene exchange between populations. The results are in best agreement with a model in which this gene exchange occurred some time in the past rather than recently.
It is important to note that such gene exchange does not need to be very large before it overwhelms separation time as a factor determining population divergence in LD values. The conclusion regarding gene exchange from the present paper is similar to the proposal of multiple migration and back-migration events suggested by Templeton (Reference Templeton2002) from single locus analyses. The time frames considered in the present study are, however, much shorter. The calculations of Fig. 4 are based on the assumption of an actual separation time of around 0·06 Myr, as opposed to separation times suggested by Templeton ranging up to and beyond 1 Myr. In reconciling these estimates, it therefore needs to be borne in mind that the LD analysis of the current paper is very sensitive to low levels of migration. Once migration has been invoked as a factor in limiting the divergence of LD values, the possibility of separation times much longer than 0·06 Myr cannot be ruled out.
I am grateful for the advice and encouragement of Bill Hill over a number of years in discussions of LD theory, particularly in the writing of the present paper. I am also grateful to Eugene Seneta for his suggestions on possible alternative approaches. Hidenori Tachida, Peter Visscher, Maria Luisa Castro and an anonymous reviewer made valuable suggestions for improvements in the paper.




