



Burkholderia pseudomallei is a select agent (Tier 1) as classified by the US Centers for Disease Control and Prevention (CDC), which can infect both animals and humans and causes the disease melioidosis [Reference White1]. Melioidosis was generally thought to be endemic in Southeast Asia and Northern Australia but is now known to be distributed on a much wider scale [Reference Tellapragada2–Reference Doker5]. Clinical manifestations of melioidosis vary greatly, from pneumonia to sepsis, with a high mortality rate of 40%, and a clinical relapse rate up to 20% [Reference Limmathurotsakul6–Reference Fang8]. Hainan is the main melioidosis-endemic area of China and covers an area of 33 210 km2 with a population of over 9 million residents [Reference Fang8–Reference Li, Lu and Han10].
Multilocus sequence typing (MLST) has been used for the molecular epidemiological study of B. pseudomallei since 2003, and MLST data have proved to be easy-to-use, unambiguous and readily comparable across laboratories [Reference Fang11]. To date, approximately 1453 sequence types (STs) of B. pseudomallei have been identified worldwide, and the number of new cases identified by MLST increases each year. We have adopted this methodology as a standard analysis strategy for all B. pseudomallei strains isolated from cases of melioidosis in Hainan and previously published a phylogenetic and epidemiological study on 98 such isolates (2002–2014) [Reference Fang11]. Here, we report further phylogenetic analysis of 52 strains from cases in the province typed by MLST in the last 3 years to examine their genetic stability over time.
Fifty-two B. pseudomallei strains were isolated from melioidosis cases between January 2014 and August 2017 in Hainan Island; two were first isolated from non-coastal cities (Baisha and Qiongzhong). Most isolates were recovered from blood (63.5%) and pus (34.6%), others were from sputum (3.8%) and urine samples (3.8%) (Table 1). Clinical samples were cultured on Columbia blood agar incubated at 37 °C for 2–3 days. B. pseudomallei identification was confirmed by the Vitek 2 Compact system (BioMerieux, Missouri, USA), and 16S rRNA PCR as previously described [Reference Fang11].
Table 1. Properties of the 52 strains of B. pseudomallei strains studied

a The numbers in bold represent the novel STs, the death cases are labelled with underscore.
b The shaded regions represent the regions which were identified as isolation locations in Hainan Island.
For MLST, PCR amplification of seven housekeeping genes was performed as previously published [Reference Fang11]. The sequence data for each allele were trimmed to a determined length and defined as relative allele numbers according to the B. pseudomallei MLST database (https://pubmlst.org/bpseudomallei). STs were assigned and all strain numbers were deposited in the database.
Clinical characteristics and patient demographic data were analysed in Microsoft Excel 2016. The phylogenetic relationships of all strains were generated using e-BURST v3 and compared against all strains in the database with JAVA 8.0. The STs of 98 strains previously described from Hainan [Reference Fang11] were also analysed to show the relationship and mutational trend between the two groups of strains. The interactive tree of life (iTOL) v3 (https://pubmlst.org/bigsdb?db=pubmlst_bpseudomallei_isolates) was used to display phylogenetic relationships. This tool generates neighbour-joining trees from concatenated nucleotide sequences based on the pair-wise differences in the allelic profiles of strains [Reference Letunic and Bork12].
The distribution of melioidosis cases across the 17 city areas of Hainan Island between the two surveys 2002–2014 (black) and 2014–2017 (red) is shown in Supplementary Fig. S1. The ages among the 52 more recent cases ranged from 1 to 79 years (median – 47 years); the majority were male (76.9%) and farmers (71.2%). Pneumonia (71.2%) and sepsis (13.5%) remained the major manifestations of melioidosis, with some cases presenting with abscesses (5.8%) and soft tissue infections (7.7%). Seven deaths occurred following pneumonia or sepsis, and old age (⩾58 years); two of the deaths were associated with novel STs (Table 1).
All 52 strains were resolved into 22 STs, five of which (ST1480, ST1481, ST1482, ST1483 and ST1484) were novel (Table 1). The dominant STs occurring in ⩾4 cases were ST50 (seven cases; 13.5%), ST58 (six; 11.5%), ST46 (five; 9.6%) and ST658 (four; 7.7%); these four STs accounted for 42.3% of all cases and the remainder were associated with ⩽3 cases. This distribution reflects that found in the first survey where the dominant STs were ST46 (13 cases; 13.2%), ST50 (11; 11.2%) and ST58 (11; 11.2%), accounting for 35.7% in all 98 strains of B. pseudomallei.
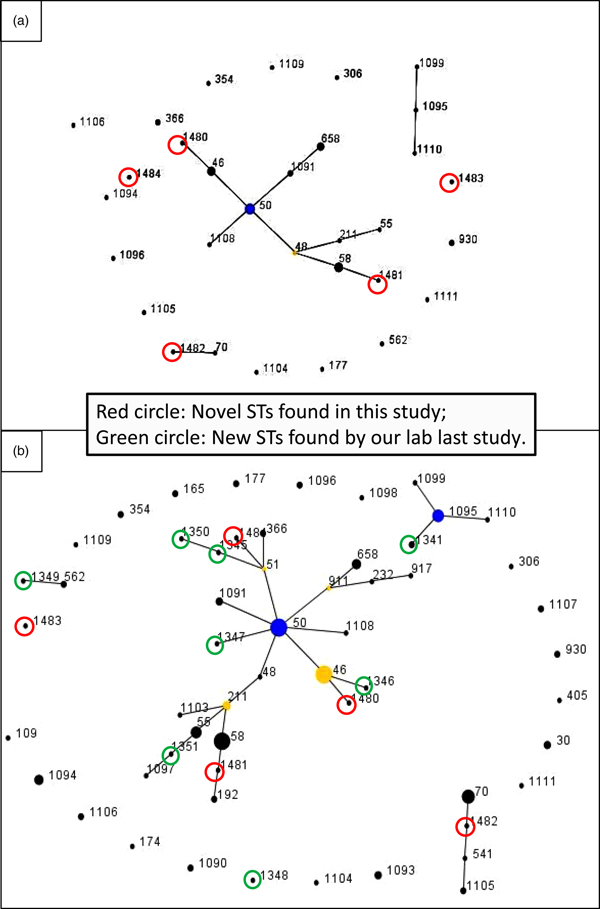
The pair-wise differences in the allelic profiles of the STs identified in both strain collections are shown in Fig. 1a and b with novel STs highlighted by red circles. The three dominant ST48, ST50 and ST46 were grouped into a single clonal complex (CC), and two novel ST1480 and ST1481 fell in the same CC (Fig. 1a) with other novel STs in individual CCs. ST1482 was phylogenetically related to ST70 which originated from Thailand. Figure 1b shows the evolutionary trends and phylogenetic relationships between STs identified in the two studies (from 2002 to 2017). Novel STs identified in each study period are highlighted in green (2002–2014) and red (2014–2017), respectively. Half of the cases were clustered into a single dominant CC, with ST50 as founder, and ST46 as sub-founder. Likewise three of the novel STs in the present study (ST1480, ST1481 and ST1484) and five novel STs from the first study (ST1345, ST1346, ST1347, ST1350 and ST1351) were grouped in the same CC (Fig. 1b). Furthermore, some novel STs originated from the same founders but generated different gene types: ST1484, ST1345 and ST1350 (founder: ST51), ST1480 and ST1346 (founder: ST46), ST1481 and ST1351 (founder: ST211) (Fig. 1b). Additionally, ST1482 and ST1483 were grouped in different CCs; ST1482 showed close linkage to both ST70 and ST541, both of which were first isolated from Thailand and ST1483 had a relatively close relationship with ST1349, an ST related to ST562, which was shared by Australian and Chinese strains of B. pseudomallei [Reference Li, Lu and Han10].

Fig. 1. e-BURST analysis of the B. pseudomallei strains. (a) MLST data of 52 STs in this study. The primary founder (yellow), ST48, is positioned centrally in the cluster and subgroup founder (blue) is ST50. Strain groups are connected by dark lines and the five novel STs are highlighted by red circles. Dot diameter reflects the number of cases. (b) e-BURST of sequence types for combined 150 strains from both surveys. Novel STs highlighted by red circles and green circles, respectively. Dot diameter reflects the number of cases.
The neighbour-joining tree shows the phylogenetic relationship among the 22 STs identified in strains from this study (Fig. 2). The five novel STs were genetically close to ST1105, ST1099, ST55 and ST58, and ST1096 (all first isolated from China, except for ST58, which was first isolated from Thailand). The dominant allele profiles were mostly the same as previously found, except for ndh-3; allele ndh-1 predominated in the first strain collection. Some rare alleles were evident between the studies, such as gmhD-36 (ST1196) and lepA-68 (ST1108) (Supplementary Table S1).

Fig. 2. Interactive tree of life (iTOL) of MLST data from the present study. Neighbour-joining trees from concatenated nucleotide sequences based on the pair-wise differences in the allelic profiles. Twenty-two STs (identified from 52 strains, five novel STs labelled with asterisks).
It is clear that B. pseudomallei is no longer restricted to tropical regions but is increasingly found in non-endemic areas [Reference Limmathurotsakul6]. Melioidosis is relatively uncommon in China but most reported human cases in the country are from the tropical Hainan island [Reference Fang8–Reference Li, Lu and Han10], and as in other countries, pneumonia remains the most common presentation of the disease [Reference McLeod7].
MLST has been repeatedly shown to be a simple and shareable strategy for molecular epidemiological studies of B. pseudomallei in various regions [Reference Tellapragada2, Reference Fang11]. Previously, we published an MLST study of 98 B. pseudomallei strains from Hainan, and here we have characterised 52 recently isolated strains and identified five novel STs. An e-BURST map of the 150 strains from both collections showed a clonal cluster with ST50 (first identified in China) as the founder genotype and that the genetic structure had remained largely unchanged over the 16-year period. Neighbour-joining tree analysis also suggests that recently emergent novel STs are closely related to some of the historical STs first isolated from Hainan Island. The linkage of two of the novel STs (1481 and 1483) to ST58, originating from Thailand, which has a close interplay with China [Reference Chen13]. Hainan attracts millions of tourists per year, which likely increases the risk of spreading infection among wider populations. A few melioidosis cases related to a history of travel in Hainan have been recorded by local hospitals in recent years (unpublished observations). In addition, the communications and commerce activities between the countries may promote the interaction of B. pseudomallei with different genetic backgrounds, and possibly give rise to the evolution of new gene types [Reference Chen13, Reference Price14]. No correlation was found between STs and geographic location, isolation source and clinical outcome of the patients but further genomic analysis may reveal hitherto unrecognised relationships [Reference Nandi15].
In conclusion, this study has confirmed the relative genetic stability of B. pseudomallei strains in Hainan over a 15-year period. With the exception of the appearance of some novel STs, the clonal populations have remained mostly conserved but further monitoring of population structures and evolutionary trends through cooperative research with other endemic areas in the region is warranted.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0950268818002741.
Acknowledgements
We are grateful to Dr Erin Price (the curator of the B. pseudomallei MLST database, Royal Darwin Hospital Campus, Australia) for useful suggestions during the analysis.
Financial support
This work was supported by the National Natural Science Foundation of China (grant numbers 81772141 and 81471914).
Conflict of interest
None.
Ethical standards
This study was approved by the Human Research Ethics Committee of the Third Military Medical University, which is a member of the Chongqing City Ethics Committees of China. All clinical cases were anonymised without personal information.





 Open access
Open access