



Introduction
Living organisms have two key barriers against possible harmful invaders: the biological barrier, such as the skin, and the immune barrier. In humans, it is possible to distinguish the latter between innate and adaptive immunity. Innate immunity is the host's first line of defense against microbial infectious agents. During the antiviral response, the innate response is responsible for quickly detecting and defending against invaders, while creating a bridge to the adaptive response [Reference Bedford1].
Mammalian antigen presenting cells (APCs), such as macrophages and dendritic cells (DCs) are able to detect microbes through pattern recognition receptors (PRRs), most notably Toll-like receptors (TLRs), retinoic acid inducible gene I-like receptors (RIG-I), such as RIG-like receptors (RLRs), the nucleotide-binding oligomerisation domain (NOD)-like receptor family proteins (NLRs) and absent in melanoma 2 (AIM2). Through PRRs, such as mainly AIM2 and NLR in contact with pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns are able to induce the formation of the inflammasome (a large multiprotein complex that is responsible for processing pro-inflammatory cytokines and creating membrane pores), which leads to a cell death called pyroptosis [Reference He2–Reference Shi4].
The innate immune system creates interferons (IFNs) and cytokines essential for elimination of viral agents. However, innate immune signalling must be well regulated, as over-activation can lead to systemic inflammation and tissue damage to the host [Reference Weston and Frieman5].
Coronaviruses are a group of single-stranded, positive-sense viruses belonging to the family Coronaviridae and subfamily Orthocoronavirinae that are largely responsible for common colds in humans. The family Coronaviridae is subdivided into Torovirinae and Coronavirinae which contains the genera Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus. However, in turn, in December 2019, there was the first record of coronavirus disease 2019 (COVID-19) in Wuhan, China. The aetiological agent is the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), of the genus Betacoronavirus, of zoonotic origin has perpetuated itself around the world causing a pandemic announced in March 2020 by the World Health Organization (WHO), which is already reported to be the cause of more than 3 million deaths and more than 150 million ill. Infected senior citizens with comorbidities had the worst prognosis [Reference Rabaan6].
COVID-19 is a highly contagious severe respiratory disease. It has been shown to be more infectious than other coronavirus types that have created outbreaks in humans, such as SARS-CoV and Middle East respiratory syndrome (MERS-CoV), with several viral strategies for evading the host immune response. Some of them are involved in inhibitory devices of the innate immune response [Reference Fung and Liu7].
This family of viruses has in particular a long RNA strand and a peculiar replication strategy. SARS-CoV-2 has an RNA strand of about 29.9 kb in length. Like other coronaviruses, this virus has at least six extra open reading frames (ORFs) in its genome. The first ORFs (ORF1a/b) are about two-thirds of the entire genome length and encode 16 non-structural proteins closer to the 5′ end (nsps 1–16). These ORFs produce two polypeptides, including p1a and pp1ab. The other one-third of the genome corresponds to structural proteins already present near the 3′ region [Reference Guo8].
SARS-CoV-2 has four structural proteins, known as S (spike), E (envelope), M (membrane) and N (nucleocapsid) proteins and the genes for accessory proteins (protein (HE), 3, 7a, among others). The S protein binds to the human ACE2 receptors (angiotensin-converting enzyme 2, a transmembrane protein), transferring the genetic material to the cell interior, initiating the replication process. After binding to the receptor, the initiation of protein S is mediated by transmembrane serine protease 2 (TMPRSS2), a transmembrane serine protease of the host cell, which involves cleavage of protein S at the sites S1/S2 and S2 [Reference Sette and Crotty9].
Physiologically, the typical RAS pathway consists of a series of steps that ultimately catalyse the formation of angiotensin (Ang) II, which promotes vasoconstriction, sodium reabsorption and water retention to elevate blood pressure. Ang II causes these effects by binding to and activating the Ang II type 1 receptor (AT 1R). Subsequently, the ACE2 axis, an anti-regulatory arm of the RAS, was detected. ACE2 inactivates Ang II by converting it to Ang 1–7, a peptide with anti-inflammatory, antifibrotic and vasodilatory properties thanks to activation of the Mas receptor (MasR). Therefore, ACE2 can act as a receptor for SARS-CoV-2, but has a protective effect on the RAS pathway [Reference Viveiros10].
In most patients, the recruited cells destroy the pathogen and then the immune response is reduced in a controlled manner. However, in some cases, this cellular recruitment brings about lung infiltration by immune system cells, triggering a hyperinflammatory response caused by exacerbated cytokines, which is a determinant grievance for patients with comorbidities, since as already mentioned it can cause tissue damage and even systemic, being a predictive factor for death [Reference Safwat11].
In this context, the following research problem arises: ‘What immunopathological aspects of SARS-CoV-2 in innate immunity are involved in the progression of COVID-19?’ The aim of this paper is to answer the problem and to build a didactic model of the innate immune response in COVID-19.
Methods
Study design
This is a systematic review to collect concise, complete and recent data on innate immunity in COVID-19. The PRISMA flowchart tool that is part of the Preferred Reporting Items for Systematic Review and Meta-Analysis Protocols (PRISMA protocol) was used to show how the final sample was arrived at, describing all the steps, inclusions and exclusions.
The study followed the following training steps: (1) preparation of the guiding question; (2) stipulation of inclusion and exclusion criteria; (3) choice of articles; (4) analysis of articles (5) interpretation, discussion and presentation of the review [Reference Rother12]. To create the guiding question the PICO strategy was used, related to the following categories: Population; Intervention; Comparison; Outcome [Reference da Santos, de Pimenta and Nobre13]. In this way, the question is described by: Population, are patients with COVID-19; Intervention is about evaluating the immunopathogenic aspects in patients with SARS-CoV-2 infection; Comparison is to relate the immunopathogenic aspects of the virus and innate immunity; Outcome resides in disease progression. Then, it resulted in: ‘What immunopathogenic aspects of SARS-CoV-2 in innate immunity are involved in the progression of COVID-19?’
Data sources
The search terms that were used for the search based on the Medical Subject Headings (MeSH) are: ‘SARS-CoV-2’; ‘COVID-19’; ‘Innate Immunity’; ‘Immunity’; ‘Cytokines’. Articles were searched with the combination of the descriptors and Boolean operator ‘AND’. The following databases were used in the literature search: National Library of Medicine National Institutes of Health of the USA (PUBMED), Latin American and Caribbean Literature in Health Sciences (LILACS), Medical Literature Analysis and Retrieval System Online (MEDLINE) and Scientific Electronic Library Online (SCIELO).
Study eligibility criteria and participants
Articles published from February 2020 to July 2021, available complete, original, narrative reviews, systematic reviews, clinical trials, in vitro assays, quasi-experiments, ecological studies, comparative studies, cross-sectional studies, case series, case-controls, cohort studies (prospective and retrospective) and meta-analyses were defined as inclusion criteria, and could be in English or Spanish. The exclusion criteria were articles published before 2020 and after July 2021, articles with only abstracts available, letters to the editor and articles or materials with topics not pertinent to the research question. The participants of the study were people infected by the SARS-CoV-2 infection.
Quality assessment and risk of bias
Two authors (MJAS and YCR) independently used the version 2 of the Cochrane risk-of-bias tool for RCTs (RoB 2), Joana Briggs Institute (JBI) Critical Appraisal (for the review articles) and the Risk of Bias in Non-randomised Studies of Interventions (ROBINS-I) tools for evaluating the methodological quality and the risk of bias of the studies [Reference O'Neil14–Reference Sterne JA and Higgins JPT et al.16]. Any discrepancies during the process were ironed out with the help of a third author (KVBL).
Data collection and extraction
Two researchers (MJAS and YCR) independently extracted data from the included publications in accordance with a predefined procedure. The same two researchers (MJAS and YCR) also assessed and organised the data in Microsoft Office Excel 365, collecting the following information: (1) title; (2) database; (3) methodology; (4) results relevant to the research topic. These observations have been represented below in a tabular form. Any discrepancies were ironed out with the help of a third author (KVBL).
Results
With the application of the inclusion criteria, a total of 102 articles were obtained; however, some articles were letters to the editor, they were not available in full or provided information that was not relevant to the research question (Fig. 1). Thus, the final framework of articles consisted of 53 articles (Table 1). The articles were entirely international (53) derived from PUBMED, LILACS, MEDLINE and SCIELO. The didactic model was constructed based on the results achieved by the bibliographic search in Table 1 (Fig. 2).
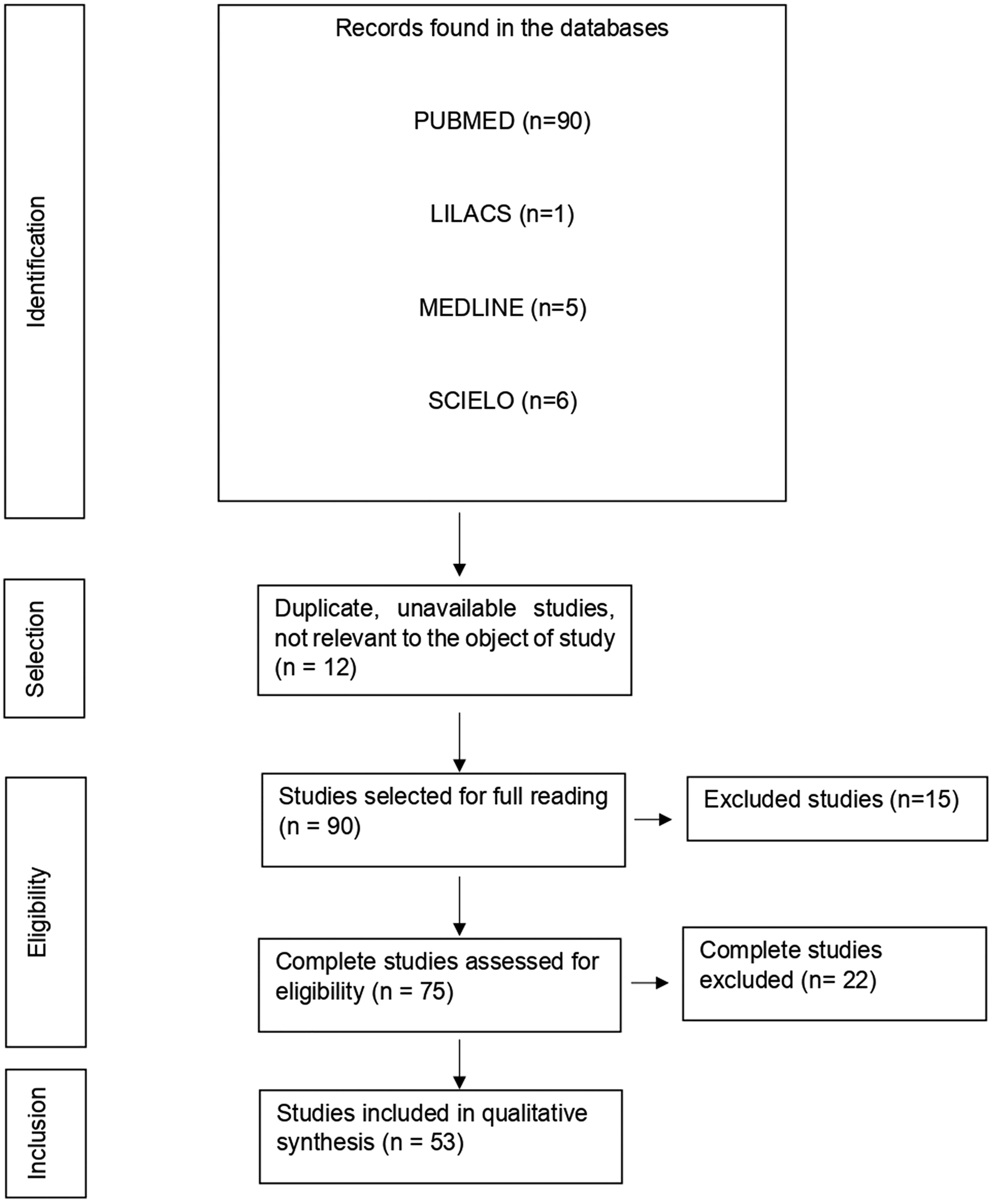
Fig. 1. Flowchart of procedures for identification, selection, eligibility and inclusion of studies for analysis. Belém, PA, Brazil (2021).

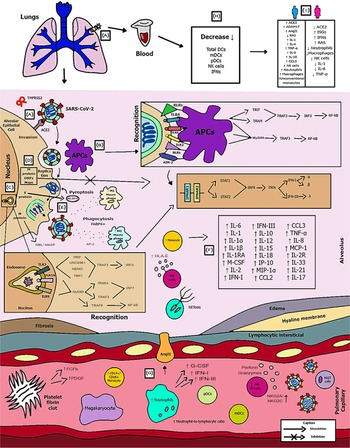
Fig. 2. Schematic model of innate immunity in SARS-CoV-2 infection.
Table 1. Characteristics of the studies included in the systematic review
Figure 2 shows the (A) entry of the virus through the alveolar epithelial cell through the binding of spike protein (S) with ACE2 cleaved by TMPRSS2.
(B) It causes the recognition of APCs through their PRRs, such as, primarily AIM2, NLRs, RLRs, TLRs (TLR2, TLR4 and TLR6) that identify the PAMP of SARS-CoV-2. TLR pathways recruit important downstream adapter proteins, such as TRAFs, MyD88, interferon regulatory factors (IRFs) and nuclear factor-kappa B (NF-κB) for induction of IFN-I (driven by IFNAR1 and IFNAR2), IFN-III.
(C) Inside the endosomes, TLR3, TLR7/8 and TLR9 operate as PRRs recruiting important downstream adapter proteins to recognise the PAMPs from the virus and stimulate the IFN response.
(D) The virus generates immunological dysregulation through its replication and maturation, in which, through the host cell, it produces structural and non-structural proteins. The membrane (M) inhibits the generation of IFN-I and IFN-III. Regarding the non-structural, Nsp proteins play a major role in virulence, such as Nsp1, Nsp3, Nsp12, Nsp13, Nsp14, Nsp15 and Nsp16, which have shown to have a potential role in the inhibition of IFNs. Furthermore, ORFs such as ORF3, ORF3a, ORF3b, ORF4a, ORF4b, ORF6, ORF8 and ORF9b inhibit TLR pathways such as NEMO and IRFs and IFN-signalling pathways.
(E) The envelope (E) induces NLRP3 inflammasome activation that generates the pyroptosis and the nucleocapsid (N) activates the complement lectin pathways (MBL and MASP), which play a pro-inflammatory role in the patient.
(F) The pathogenesis of the virus can cause fibrosis, oedema, thrombotic events, lung damage and a high inflammatory process. Pro-inflammatory cytokines and chemokines, such as interleukin-1 (IL-1), IL-1α, IL-1β, IL-2, IL-1RA, macrophage colony stimulating factor (M-CSF), granular colony stimulating factor (G-CSF), IL-6, IL-10, IL-12, IL-15, IP-10, macrophage inflammatory protein 1-alpha (MIP-1α), chemokine ligand 3 (CCL3), tumour necrosis factor-alpha (TNF-α), IL-8, IL-18, CCL2, IL-2R, IL-33, IL-21 and IL-17 have been associated with disease severity.
(G, H) In severe cases of COVID-19, innate immune cells such as macrophages are increased in mixed polarisation M1/M2 (with FABP4+ phenotype, which is associated with a persistently hyperinflammatory phenotype profile in a hyperactivated state), there is induction of CD14+ and CD16+ monocytes with lower expression of HLA-DR (defining an acquired immune-suppression), reduction of natural killer (NK) cells (with different immunotypes and higher expression of HLA-E, NKG2A and NKG2C, causing overactivation of NK cells), high recruitment of neutrophils with production of neutrophil extracellular traps (NETs) that create a situation of NETosis in the alveoli, high level of neutrophils in proportion to lymphocytes, reduction of T cells of the mucosa and of total DCs (from plasmacytoid dendritic cells – pDCs and mature dendritic cells – mDCs). Platelets stimulate growth factors such as FCFb and PDGF that promote cellular proliferation, collagen production and other elements of the cellular matrix, enhancing the healing process, even under adverse conditions. pDCs and macrophages show a complete response to SARS-CoV-2, launching a late but robust IFN type I response and releasing other inflammatory cytokines against the virus. Both type I and type III IFNs are induced at high levels in pDCs upon SARS-CoV-2 stimulation.
(I) Innate immunity seems to affect and protect the sexes differently, due to several factors, such as possibly increased production of the activate disintegrin and metalloproteinase (ADAM17) gene in men, reduction in the typical RAS pathway, increased activation of angiotensin II (AngII or Ang1–7), high recruitment of neutrophils, NK cells, monocytes and macrophages and, in women, a lower level of ACE2, which allows greater generation of interferon-stimulating genes (ISGs), with more IFNs, with a lower inflammatory picture of cytokines in females, which despite that, is more effector in males, with production of IL-8, IL-18, CCL5 and unconventional monocytes.
Discussion
Despite multifaceted efforts, certain treatments for SARS-CoV-2 have shown controversial benefits [68–72]. These include remdesivir, immunoglobulins, monoclonal antibodies to protein S and anti-inflammatory agents. New vaccines are alternatives to stop the progression of the pandemic. However, in the early stages of the outbreak, attention should be focused on treatments designed to control the cytokine storms that occur in patients with SARS-CoV-2 pneumonia. Efforts have been made to control the components of innate immunity that may contribute to the morbidity and mortality of SARS-CoV-2 pneumonia. Thus, understanding the structure of novel viruses and their interactions with the immune system is essential for drug and vaccine production.
According to Mason, the pathogenesis of COVID-19 in 2020 can be divided into three stages: the first stage is asymptomatic and occurs when the inhaled SARS-CoV-2 virus begins to replicate in nasal epithelial cells; the second stage occurs when the virus passes through the lungs, continues to spread, and triggers a stronger innate immune response and the third stage manifests as hypoxia, ground-glass infiltration and development of respiratory failure [Reference Mason73]. Clinical presentations range from no symptoms to mild fever, cough and dyspnoea to cytokine storm, respiratory failure and death. The mechanisms used by CoV to drive IFN responses can be divided into three categories: evasion (the virus protects itself from PRR), as a strategy to drive dsRNA replication (produced as a mediator of transcription) in double-membrane vesicles; inhibition of IFN induction by the virus, which inhibits IFN transcription; inhibition of IFN signalling with the help of its eight specific viral proteins that suppress IFNAR signalling (IFNα/β receptor) [Reference Taefehshokr65].
SARS-CoV-2 appears to act on the activation and maturation of IL-1β, which in turn activates other pro-inflammatory cytokines, such as IL-6 and TNF-α. The immunopathology of COVID-19 is characterised by an elevation of IL-6 and TNF-α. These cytokines are products of activation of the TLR4, which is part of innate immunity. IL-1 levels are related to the virulence and severity of the infectious process [Reference Costela-Ruiz74].
IL-6 contributes to the host's defense against infections and tissue injury. However, exaggerated and excessive synthesis of IL-6 accounts for its elevated serum levels in COVID-19 patients and is associated with disease severity due to its ability to recruit different cell immunotypes in innate immune response. Patients infected with COVID-19 harbour an expanded population of circulating monocytes that secrete IL-6 and IL-1β, as a result, patients with COVID-19 have elevated serum IL-6 as well as lactate dehydrogenase levels compared with healthy controls [Reference Chen75]. TNF-α is generated by macrophages, monocytes, endothelial cells, neutrophils, smooth muscle cells, activated lymphocytes, astrocytes and adipocytes. It is associated with the disease severity by sustaining inflammatory milieu and plays a role in the formation of oedema [Reference Fara76].
In vitro stimulation by IL-6 and soluble IL-6 receptor previously revealed impaired cytolytic functions (perforin and granzyme B production) by NK cells from healthy donors, which can be restored after addition of tocilizumab (IL-6R blockade) [Reference Huang48]. Although it is the main cause of cytokine storms in COVID-19 patients, IL-6 has both anti-inflammatory and pro-inflammatory properties that play a complex role in COVID-19 pathology.
Upon virus entry, SARS-CoV and SARS-CoV-2 activate disintegrin and metalloproteinase (ADAM17), which are cellular events that help differentiate mild and severe coronavirus infections. Importantly, activation of the standard RAS pathway (ACE/AT 1R) resulted in an increase in ADAM17 activity. Therefore, hyperactive acute respiratory distress syndrome (ARDS) promotes disease severity, especially in patients with cardiovascular diseases as comorbidities [Reference Viveiros10].
Innate immune cells appear to be responsible for the inflammatory environment in the lungs. Cytokine storms are a complex network of interactions between immunologically important molecules and molecular events expressed in the clinical phenotype of systemic inflammation, multiple organ failure and increased blood potassium. This framework releases large amounts of pro-inflammatory cytokines, as in COVID-19, including IL-6, TNF, IL-1β, IL-12, IL-17, IL-18, IP-10, IFN-γ, CCL2 and CCL5 [Reference Abers43, Reference Azkur77]. In addition to further stimulating the cytokine storm, SARS-CoV-2 was able to infect DCs and limit DC maturation, thus suppressing T-cell responses. Three studies demonstrated a high proportion of CD14+ and CD16+ cells in patients with severe COVID-19 [Reference Zhou46, Reference Zhou78, Reference Zhou79], with inflammatory factors including GM-CSF and IL-6, IL-10 and TNF-α. Alveolar FABP4+ macrophages predominate in severely damaged lungs. The alveolar FABP4+ macrophage induces viral inflammation because it produces high levels of IFN and chemokine-stimulated genes [Reference Ricci21, Reference Liao80]. These results suggest an association between elevated levels of GM-CSF and IL-6 secreted by monocytes and cytokine storm.
The IFN-mediated antiviral pathway is one of the primary mechanisms of innate immunity against invading viral pathogens and consists of two steps, including IFN activation and IFN signalling. First, viral infection rapidly induces the expression and secretion of type I and type III IFNs. In the terminal phase, secreted type I and type III IFNs bind to cell surface receptors and initiate Janus kinase (JAK) and transcriptional activator (STAT) signalling. When IFN binds to these receptors, intracellular tyrosine kinases, including JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2), are activated and phosphorylated, resulting in increased phosphorylation and activation of STAT1 and STAT2 [Reference Mu40].
INF type I is considered the first line of defense against viruses [Reference Trouillet-Assant41]. IFN-I is a family of cytokines that bind to the IFN receptor and is composed of two transmembrane subunits, IFNAR1 and IFNAR2. The two receptors include an extracellular domain that binds to IFN-I, a transmembrane helix and an unstructured intracellular domain that binds to JAK and STAT (signal transducers and transcription activator signalling pathways) transcriptional activator signals. Type I IFN via IFNAR activates JAK and STAT. After IFNAR signalling, JAK1 and TYK2 phosphorylate STAT1 and STAT2 molecules to form a complex with IRF9 and interferon-stimulating gene factor 3 (ISGF3). Type I IFNs correspond to IFN-α, IFN-β, IFN-ε, IFN-ω and IFN-κ and the IFN-II and IFN-III families include IFN-γ and IFN-1. Consequently, IFN-α strongly inhibits SARS-CoV-2 replication in vitro [Reference Lokugamage81]. In the case of IFN-β, after ORF6 binds to its receptor it activates the JAK-STAT pathway, in which the kinases JAK1 and TYK2 phosphorylate STAT1 and STAT2, triggering their dimerisation and nuclear translocation [Reference Lei19].
These complexes are inserted into the nucleus and stimulate the transcription of ISGs, followed by the expression of antiviral proteins. IFN-stimulating genes are an integral part of the innate antiviral defense mechanism that limits viral invasion and restricts viral replication after the virus invades the host cell. Several ISG products, including IFN-inducible transmembrane proteins (IFITM) 1, 2 and 3, limit SARS-CoV-2-mediated infection [Reference Hosseini82]. From an immunological point of view, IFN-I has three main functions: to activate the antiviral state of infection and adjacent cells, limiting the spread of infection; to regulate innate immune responses, such as antigen presentation and NK cell function, limiting the inflammatory pathway; to activate the adaptive immune system to develop specific T and B cell responses to high-affinity antigens [Reference Wilmes83].
Class III IFN (IFN-λ) showed stronger antiviral function than IFN-α in treating influenza infection without activating IFN-α-induced inflammation and tissue damage. On the other hand, IFN-λ treatment has been shown to interfere with the detection of bacteria by lung neutrophils during superinfection with influenza, which alters the lower respiratory tract, weakens defenses and the risk of coinfection of patients with COVID-19 may increase. Similar to IFN-I, IFN-λ is reduced during infection with COVID-19, and IFN-λ suppresses SARS-CoV-2 replication in vitro in human enterocytes [Reference Stanifer18, Reference Major49, Reference Broggi50, Reference Taefehshokr65].
Defective type I IFN immunity was shown to underlie life-threatening COVID-19 pneumonia. Genetic defects observed in critically ill COVID-19 patients are acquired during disease evolution through secondary events, in deficient pDCs. Despite this, the specific role of DCs in the COVID-19 pathology has been insufficiently studied, so far [Reference Zhang55, Reference Greene and Zuniga84].
Thus, with reduced serum levels of DCs and plasmacytoid DCs, the combined effects of a naturally poor IFN-inducing viral strain and intrinsic defects in antiviral immune or epithelial responses within the nasal mucosa may predispose to severe disease. These occur due to increased viral replication in the upper airways. NK cells have the role of recruiting their different immunotypes to exert their cytotoxicity. Furthermore, NK cells exhibit an activated and cyclic phenotype in acute SARS-CoV-2 infection at both the protein and transcriptomic levels, with upregulation of Ki67, CD69, HLA-DR and CD38. The upregulation of inhibitory checkpoint receptors such as LAG3, TIGIT and TIM3 reduces the formation of NK cells [Reference Maucourant22, Reference Xu24].
The N protein catalytically modifies the host protein ACE2 by SUMOylation and ubiquitination, respectively, by interaction with the host proteins hUbc9 and TRIM25, respectively, as well as acting as an antagonist of IFN-I production. The circadian shift of ubiquitination towards the N protein as a RIG-I target for proteolysis, thereby redirecting cells away from critical virus detection PRRs is well described, while the target of N-mediated SUMOylation remains unclear. Elevated levels of IL-6, IL-10, TNF-α and interleukin 2 receptor (IL-2R) are associated with disease severity. In addition to manipulating cytokines, CoV also controls other immune processes, including antigen presentation [Reference Mu40, Reference Vabret85].
It has been hypothesised that the hyperinflammatory innate immune system caused by SARS-CoV-2 infection cannot be eliminated due to its antagonism of the innate immune response [Reference Birra32]. Therefore, it induces an excessive release of inflammatory cytokines and compensates for the depletion of the immune system caused by SARS-CoV-2-induced lymphopaenia [Reference Fathi and Rezaei86]. In addition, TLR3- and IRF7-dependent IFN-I innate errors of immunity have been found and related to disease severity. The most thought-provoking are the autosomal-recessive (AR) deficiencies of IRF7 and IFNAR1. The AR form of IFNAR1 deficiency highlights the importance of type I IFN production over type III IFN production, which is also impaired by defects of TLR3, IRF7 and IRF9 [Reference Zhang55].
Regarding the pulmonary aetiology, the overproduction of cytokines after SARS-CoV-2 infection increases the permeability of the capillary wall membrane around the infected alveoli, causing pulmonary oedema, dyspnoea and hypoxaemia [Reference Meftahi87]. Plasma exudation and loss of alveolar elasticity due to reduced surfactant production owing to SARS-CoV-2 infection of type 2 lung cells induces ARDS in patients with COVID-19. Following cytokine storms, these SARS-CoV-2-induced immune disturbances can lead to increased microbial infections, septic shock and severe multiple organ failure [Reference Yang47].
Another important pathological consideration in patients with SARS-CoV-2 pneumonia is their propensity for thrombotic events. Alternative and lectin pathways are effector mechanisms of innate immunity. Many symptoms can result from activation of the complement system by alternative and likely lectin binding pathways. This includes the tendency to ARDS and the potential for hypercoagulability. Strong complement activation can induce activation of the coagulation system (with the appearance of thrombotic events in endothelial vessels). Therefore, inhibition of complement activation may prevent thrombotic complications of SARS-CoV-2 pneumonia [Reference Jordan67].
TLR agonists can also be used as prophylaxis for SARS-CoV-2. Proud et al. [Reference Proud88] demonstrated that prophylactic use of a TLR2/6 agonist reduces SARS-CoV-2 infection and protects against COVID-19. TLR2 stimulation leads to activation of the innate immune response, suppression of excessive inflammation and tissue damage, as well as generating the integrity of local epithelial barrier function. TLR promotes the recruitment of Toll-interleukin (TIR) domain-containing adaptor proteins, such as myeloid differentiation factor 88 (MyD88), TIR domain-containing adaptor protein (TIRAP) and TIR domain-containing adaptor protein inducing IFN-β-related adaptor molecule (TRIF), leading to activation of the transcription factors IFN regulatory factor-3 (IRF3), IRF7 and nuclear factor kappa enhancer light chain B activated cells (NF-κB) required for the transcriptional induction of antiviral IFN, pro-inflammatory cytokines and chemokines [Reference Ricci21, Reference Proud88].
In particular, activation of the IL-6 pathway by SARS-CoV-2 infection can induce vascular endothelial growth factor (VEGF), fibrinogen and factor VII. IL-6 blockade or reduced induction may be a new treatment strategy for critically ill patients with COVID-19 [Reference Bikdeli89, Reference Whyte90]. Ang II also stimulates the expression of TFs (tumour inflammatory factor mediators) in affected cells, increases thrombin formation and impairs fibrinolysis [Reference Miesbach91].
Other anti-inflammatory agents were also tested in their effectiveness by repurposed drugs for COVID-19 therapy and involved positive results under mediators such as inhibitors of C5, IL-1, JAK1, JAK2 and IL-33 and related to reducing release of TNF-α and IL-1β [Reference Kunnumakkara92].
Since thrombosis is one of the physiologically related events caused by continuous activation of the Ang II signalling pathway, ACE2 deficiency was stimulated by the pro-inflammatory condition caused by SARS-CoV-2 and may exacerbate thrombosis. It is unclear whether IFN-I therapy is an effective benefit for patients with COVID-19, since SARS-CoV-2 may use ISG to increase infectivity, and it is not known whether the IFN response limits SARS-CoV-2 replication. However, some clinical studies have shown positive data on IFN-I as a form of treatment or prevention of COVID-19 [Reference Davoudi-Monfared93–Reference Zhou97].
IFN levels and duration in a variety of human diseases can be associated with determining macrophage/monocyte resistance. Induction or suppression of tolerance is mediated by transcriptional, epigenetic and metabolic reprogramming [Reference Netea98], which leads to dysregulation of the inflammatory response. Therefore, dramatic epigenetic and metabolic changes, possibly associated with macrophage resistance and innate immune memory have recently been reported in patients with COVID-19 [Reference Ricci21].
Disruption of the immune system is one of the consequences of COVID-19. However, there are differences in the immune system between mild and severe conditions. In mild cases, the innate immunity seems to be maintained. On the other hand, in severe cases, the innate systemic process is affected. Congenital lymphoid cells (ILCs containing NK cells) have no specific antigen receptors, but are sometimes highly reactive to specific pathogens or are referred to as ‘trained immunity’ [Reference Mantovani and Netea99].
The number of NK cells and DCs (total and activated) decreased in severe cases. The number of cells expressing HLA-DR is significantly lower in severe cases, indicating that antigen-presenting cells are unable to induce an adaptive response. The virus also affects the monocyte population. The proportion of intermediate and unconventional monocytes has decreased, indicating a decrease in functional maturity. An increased proportion of cells co-expressing M1 and M2 monocyte markers suggests the development of fibrosis as a mechanism of prolonging and repairing inflammation, potentially damaging the lung parenchyma and increasing the risk of worsening clinical outcomes [Reference Matic58].
Gender differences in COVID-19 are reflected in the initial treatment of viral infections, various hormonal signalling pathways and risk status based on social and cultural factors. The differences in immune functions between women and men, which are manifested not only by stronger immune responses in women against pathogens and vaccines, but also by greater susceptibility to autoimmune diseases. Many viruses are affected by oestrogen at the molecular level, especially in the mechanism of virion replication and maturation, but so far there is no evidence for SARS-CoV-2 [Reference Viveiros10].
The literature suggests that the ability of women to induce an early innate immune response is increased and the typical trend towards increased RAS activity in diseases that are strongly dependent on both genetics and sex hormones is reduced. Thus, these candidate pathways and molecular mechanisms provide a multifaceted account of the severity and mortality of COVID-19 in men compared to women. Positive regulation of the ACE2 axis, Ang 1–7 MasR or ADAM17-mediated blockade of ACE2 release, provides an altered pathway essential for ameliorating the increased severity in men [Reference Viveiros10, Reference Takahashi59–Reference Ji62].
Furthermore, defects in the innate immune cells response to SARS-CoV-2 caused by ageing are related to poorly managed viral multiplication and inflammation during the early symptom phase and in the following illness development. These defects in the effect of age (which affects older people more compared to adults and children) are related to reduced activation of monocytes (as a consequence of downregulation of antigen presentation molecules, e.g. HLA-DR) that act as APCs and antiviral mediators, DC dysfunction (that should regulate the initial response against the pathogen) and reduced percentage of cytokine-producing NK cells (as a result of decreased expression of functional immune co-stimulatory molecules). Hence, an underlying pathophysiological reason for the lack of virological control and higher risk of disease progression in older COVID-19 patients is impaired innate cellular responses during the early stages of infection [Reference Cham100].
Conclusions
To date, SARS-CoV-2 remains prevalent as a major global health emergency due to its pathogenesis. Its immunopathogenesis reveals several immune evasion factors, which are crucial for replication, maturation and invasion into host cells that are determinants for the success of viral activity. Innate immunity appears as indispensable in the initial stage to provide the influx of combined innate immune cells, IFNs (mainly types I and III), cytokines and chemokines, and complement that regulate the longest survival of patients in pictures differentiated from mild, moderate to severe. Thus, the severity of the disease goes through innate errors, virulence of the virus and gender differences, which also correlate with comorbidities. Moreover, it is believed that much remains to be revealed about the real impact of innate immunity and the clarification of controversial theories of both prognosis and therapy on the means of this immunity.
Author Contributions
MJAS was responsible for the conceptualization, formal analysis, data curation, methodology, investigation, validation, visualization, roles/writing – original draft, editing. YCR was responsible the supervision, visualization, writing – review and editing. KVBL was responsible for the supervision, visualization, writing – review and editing. LNGCL performed the conceptualization, investigation, project administration, supervision, visualization, writing – review and editing. All authors read and approved the final manuscript.
Conflict of interest
The authors declare no conflicts of interest regarding the publication of this paper.
Data availability statement
Data availability is not applicable to this article as no new data were created for this study.


 Open access
Open access