


INTRODUCTION
Since noroviruses (NoVs) were first identified as causal agents of viral gastroenteritis, they have been found to be the most common cause of epidemic gastroenteritis in all age groups across the globe and to be the leading cause of foodborne illness [Reference Adler and Zickl1–Reference Scallan3]. NoVs are highly infectious and constitute a major cause of gastroenteritis-related hospitalization [Reference Koopmans4]. In the USA alone, the annual disease incidence due to NoVs leads to about 570–800 deaths, 56 000–71 000 hospitalizations, 400 000 emergency department visits, 1·7–1·9 million outpatient visits, and 19–21 million total illnesses [Reference Hall5]. Furthermore, outbreaks of acute gastroenteritis due to NoVs pose a considerable economic burden as a result of healthcare costs and lost productivity [Reference Navas6, Reference Lopman7]. In the USA, the cost of hospitalization for NoV illnesses has been estimated at approximately $500 million, and the cost of healthcare and lost productivity has been estimated at $2 billion [Reference Bartsch8]. Studies to reduce the burden of NoV illness have been hindered by the lack of complete understanding of NoVs. Part of this inadequate understanding of NoVs is attributable to the lack of in vitro cell-culture systems and in vivo small-animal models for human NoVs [Reference Lee and Pang9].
The typical transmission of NoV is directly via person-to-person transmission (fecal–oral and vomit–oral routes) or indirectly through waterborne, foodborne, and environmental transmission [Reference Barclay10]. The average incubation period is 24–48 h, and symptoms include vomiting (⩾50% of cases), diarrhea, nausea, abdominal cramps, malaise, and low-grade fever. The illness usually resolves in 12–72 h but can last longer in young children, elderly people, hospitalized patients, and immunocompromised people [Reference Lee and Pang9].
NoVs belong to the genus Norovirus in the Caliciviridae family. The viral genome consists of a positive-sense single-stranded RNA about ~7·7 kb long, comprising three open reading frames (ORFs). The genome is protein-linked at the 5′-end and polyadenylated at the 3′-end. ORF1 encodes the non-structural proteins p48, NTPase, p22, VPg, 3C-like protease, and RNA-dependent RNA polymerase. ORF2 encodes the major capsid protein VP1, and ORF3 encodes the minor structural protein VP2. VP1 is a structural protein containing antigenic determinants that define strain specificity. VP1 is comprised of three domains, the N-terminal; the shell domain, S; and the protruding domain, P. The P domain, which forms the protrusions on the virus, is divided into two subdomains, P1 and P2. The P2 domain, which is located at the distal surface, is a hypervariable region that plays an important role in receptor binding and immune reactivity [Reference Hardy11]. NoVs are divided into seven genogroups (GI, GII, GIII, GIV, GV, GVI, and GVII) and are classified into over 40 genotypes. Among them, three genogroups, GI, GII, and GIV, are known to infect humans [Reference Zheng12, Reference Vinje13].
At present, GII.4-type viruses are believed to be the major cause of gastroenteritis in humans, being responsible for >70% of the outbreaks [Reference Vega14, Reference Kim15]. Although GII.21 is not a dominant type of NoV, it is consistently detected in hospitalized patients with gastroenteritis and environments in many countries [Reference Vega14, Reference Mans16–Reference Pérez-Sautu24]. In some countries, NoV GII.21 is a dominant cause of gastroenteritis [Reference Yahiro25]. In Korea, NoV GII.21 was found during the winter season of 2012–2013 [Reference Kim26].
The purpose of this study was to characterize an uncommon type of NoV GII.21 isolated from a stool sample obtained from a Korean patient with acute gastroenteritis.
MATERIALS AND METHODS
Ethics statement
The stool sample for the sequencing study was provided by the Waterborne Virus Bank (WAVA, Seoul, South Korea). Because of difficulties in tracking the exact records of the pediatric patient from the donor hospital, informed consent from the parent of the said patient could not be acquired. The Institutional Review Board reviewed and approved the use of this sample for the purpose of research, as this study does not affect the patient. All the experimental work and sample collections were supervised by the Catholic Medical Center Office of the Human Research Protection Program (CMC OHRP) of South Korea (approval number MC15EASE0117).
Specimen preparation and viral RNA extraction
One hundred and ninety-five stool samples obtained from patients who presented with fever and diarrhea from January 2013 to December 2013 were screened from the WAVA. Most stool samples were from hospitalized infants (⩽3 years old), and the GII.21 sample used in this study was also from an infant patient (1 year old). The NoV-positive GII.21 sample obtained from the WAVA was stored at −70 °C until RNA extraction. The frozen stool sample was thawed and diluted with 10% phosphate-buffered saline, after which it was centrifuged. NoV RNA was extracted from the supernatant using a QIAamp Viral RNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Isolated RNA was used as a template for reverse transcription–polymerase chain reaction (RT-PCR) and stored at −70 °C until further use.
RT–PCR
For the detection of NoV-positive samples, RT–PCR was performed with the OneStep RT–PCR kit (Qiagen) using GII-F1M and GII-R1M primers (Table 1). To analyze the whole-genome sequence of NoV, nine more primer pairs were newly designed on the basis of the YO284 strain (GenBank accession number KJ196284) (Table 1). RT–PCR was performed with an S1000 thermal cycler (Bio-Rad, Hercules, California, USA), and the steps comprised reverse transcription (50 °C for 30 min), initial PCR activation (95 °C for 15 min), 30 cycles of three-step cycling (94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min), and final extension (72 °C for 10 min). All RT-PCR products were examined by electrophoresis in ethidium bromide-stained 1·5% agarose gels.
Table 1. Primers used in this study
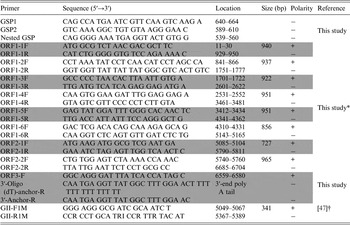
* The primers were based on the YO284 strain (GenBank accession number KJ196284).
† The primers were for NoV detection (GenBank accession number X86557).
Determination of the 5′- and 3′-ends of NoV genomic RNA
To determine the 5′-end of NoV genomic RNA, rapid amplification of cDNA ends (RACE) was performed with the 5′ RACE System for Rapid Amplification of cDNA Ends Version 2.0 Kit according to the manufacturer's recommendations (Invitrogen, Carlsbad, California, USA). Three primers (GSP1, GSP2, and nested GSP) were designed based on the ORF1 sequence for 5′-end RACE PCR (Table 1). To obtain the exact sequence of the 3′-end of the NoV genomic RNA, cDNA was synthesized using reverse transcription with 3′-oligo (dT)-anchor-R (Table 1). The second PCR was conducted using the ORF3-F and 3′-anchor-R primers (Table 1) under the following conditions: 30 cycles of three-step cycling (94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min) and 72 °C for 10 min.
Cloning and sequencing of the complete genome
All PCR products obtained were extracted using the HiYield Gel/PCR DNA Fragments Extraction Kit (RBC, Taipei, Taiwan) and were cloned into pGEM-T Easy Vectors (Promega, Madison, Wisconsin, USA). The cloned vector was transformed into Escherichia coli DH5α competent cells (RBC) according to the manufacturer's instructions and was selected from Luria–Bertani (LB) agar plates (Duchefa, Haarlem, The Netherlands) containing 40 mg/ml X-gal, 0·1 mM isopropyl-β-d-thiogalactoside, and 50 mg/ml ampicillin at 37 °C for 16–18 h. Selected clones were inoculated in LB broth (Duchefa) and incubated overnight in a shaking incubator (IS-971R, Jeiotech, Daejeon, South Korea) at 37 °C and 200 rpm. Plasmid DNA was purified using the HiYield Plasmid Mini Kit (RBC) and sequenced (Cosmo Genetech, Seoul, South Korea). The sequencing results were analyzed using BLAST (National Center for Biotechnology Information, NCBI).
Phylogenetic analysis
Comparative sequence analysis, including sequence alignments and estimation of genetic distances, was performed with Clustal W using the Molecular Evolutionary Genetic Analysis software (MEGA soft version 6.0) [Reference Tamura27]. Phylogenetic trees were constructed using the neighbor-joining method with a Kimura two-parameter model in MEGA [Reference Saitou and Nei28], and branch support was calculated based on 1000 bootstrap replicates. The complete genome sequences and partial genome sequences were collected from NCBI.
RESULTS
NoV detection and full-length genome of strain JW
Of the 195 stool samples screened, 56 (28·72%) were positive for NoV genogroup II. Only one sample (1/56; 1·79%) was a GII.21-type NoV. The full-length genome of strain JW (GenBank accession number KX079488) was 7510 nucleotides (nt) long and contained three ORFs: ORF1 (5–5104; 5100 nt), ORF2 (5085–6707; 1623 nt), and ORF3 (6707–7465; 759 nt). ORF1 and ORF2 had an overlap of 20 nt, whereas ORF2 and ORF3 had a single-nucleotide overlap. Genotyping by the online Norovirus Genotyping Tool (http://www.rivm.nl/mpf/norovirus/typingtool) [Reference Kroneman29] showed that strain JW belonged to genogroup II and genotype GII.P21 (by ORF1) or GII.21 (by ORF2). BLAST results with the full-length genome sequence of strain JW confirmed the genotyping result and showed the highest identity with strain YO284 (GenBank accession number KJ196284; query cover =99% and identity =96%). ORF1 and ORF3 BLAST results also showed the highest identity with strain YO284 (GenBank accession number KJ196284; query covers, 100% each; identities, 97% and 96%, respectively). ORF2 BLAST results showed the highest identity with strain CUHK-NS-626 (GenBank accession number KR921942; query cover =100% and identity =99%).
Phylogenetic analysis
We next attempted to understand the genetic relationship between strain JW and other published full-length genomes of NoV reference strains, which are available in GenBank. As shown in the tree, the whole-genome sequences of strain JW were classified as NoV GII.21. The full-length nucleotide sequences of strain JW showed maximum identity with those of strain YO284 (GenBank accession number KJ196284), with 96·3% similarity (Fig. 1a ). The tree based on the amino acid sequences of ORF1, ORF2, and ORF3 showed 98·6%, 96·3%, and 94·9% identities, respectively, with strain YO284 (Fig. 1b – d ).

Fig. 1. Phylogenetic analysis of NoV based on nucleotide and amino acid sequences. The neighbor-joining method in MEGA was used to construct phylogenetic trees. The numbers associated with each branch indicate the bootstrap values for the genotype. Phylogenetic trees based on (a) full-length nucleotide sequence, (b) amino acid sequence of ORF1, (c) amino acid sequence of ORF2, and (d) amino acid sequence of ORF3. Strain JW is highlighted with a solid red circle. The representative strains are named by ‘accession number/host/genogroup’ and ‘genotype/strain/collection date/country’. Country codes: CHN, China; HKG, Hong Kong; JPN, Japan; USA, United States of America; AUS, Australia; TWN, Taiwan; NLD, Netherlands; MYS, Malaysia; KOR, Korea.
Phylogenetic analysis was performed using the partial VP1 genes of strain JW and other GII.21 strains available in GenBank. Strain JW was confirmed to cluster in GII.21.b1 (Fig. 2a ) [Reference Nahar30], which comprises strains detected in water, clinical samples, and seafood. Strain JW showed a very high identity (95–97%) with other strains clustering in GII.21.b1.

Fig. 2. Phylogenetic analysis and amino acid sequence comparison of partial ORF2 gene. (a) Neighbor-joining phylogenetic analysis of the partial nucleotide sequences of the VP1 genes for strain JW (solid red circle). Strain JW clustered in GII.21.b1. (b) The comparison of the partial amino acid sequence of VP1 genes is shown as amino acid substitution patterns. The consensus sequence was based on the corresponding amino acid sequence that was most highly conserved. The substituted amino acids are in red boxes. The representative strains are named by ‘isolation source/strain/collection date/country’. Country codes: CHN, China; JPN, Japan; USA, United States of America; TWN, Taiwan; BGD, Bangladesh; THA, Thailand; IND, India; IRQ, Iraq; SGP, Singapore; NZL, New Zealand; ESP, Spain; KOR, Korea.
Alignment analysis of amino acid sequences of VP1
Figure 2b shows the comparison of partial VP1 sequences that indicate amino acid substitution patterns in S domain of VP1, which represents the most conserved region of the sequence that is used for genotyping [Reference Prasad31, Reference Kroneman32]. In the GII.21.a cluster, seven strains had the Ala36Val substitution, six strains had the Asn45Thr substitution, and five strains had the Ile47Met substitution. In the GII.21.b2 cluster, five strains had the Asn54Thr or Asn54Ser substitution (Fig. 2b ). The consensus sequence was based on the corresponding amino acid sequence of the most highly conserved sequence.
The functional VP1 protein of NoV, which functions as an antigen and determines the specific genotype, was analyzed [Reference Kroneman32, Reference Tan33]. Particularly, the P2 subdomain of VP1 (residues 279–405), is the highly variable and most exposed region of the structure as an antigen [Reference Prasad31]. Five strains were used for alignment analysis of the amino acid sequences. A total of 19 amino acid alterations (red arrow) were seen in the recently collected strains (cluster B, Fig. 3). Among these, three amino acid substitutions and one amino acid insertion (in red box) were located within the P1 subdomain, one amino acid substitution was located in the S domain, and 14 amino acid alterations were located within the P2 subdomain. A comparison of VP1 amino acid sequences of previously collected strains (cluster A) and the recently collected strains (cluster B) indicated that most substitutions were within the hypervariable region of the P2 subdomain (Fig. 3).

Fig. 3. Comparison of amino acid substitutions of five strains. Alignment of VP1 amino acid sequences of NoV strains YO284 (GenBank accession number KJ196284), Salisbury150 (GenBank accession number JN899245), CUHK-NS-293 (GenBank accession number KR921937), GL02BLPV2 (GenBank accession number AMO28394), and JW (GenBank accession number KX079488). A total of 19 amino acid alterations (red arrows) were noted in the previously (cluster A) and recently collected strains (cluster B). Most substitutions were present within the hypervariable P2 subdomain (residues 279–405).
The HBGA-binding pocket is conserved in NoV GII.21
The GII.21 lineage has major residues that form the novel histo-blood group antigen (HBGA)-binding pocket (containing the B, T, and N loops) and the conventional GII-binding interface (containing P, S, and A loops) [Reference Liu34]. The alignment of HBGA-binding pocket amino acid sequences in strain JW and three other strains showed very high identity. However, two amino acids were substituted in the N and T loops (in red box). The conserved residues forming the binding interface are shown (Fig. 4).

Fig. 4. Amino acid sequence alignment of the surface loops of the P domains forming HBGA-binding interfaces. Sequences of the three surface loops on the HBGA-binding interfaces (B, N, and T loops) representing the genotype of the GII.21 [Reference Liu34]. The conserved residues forming the binding interface are indicated with asterisks. Two substituted amino acids in the N and T loops are shown in red boxes. The representative strains were named by ‘accession number/host/genogroup’ and ‘genotype/strain/collection date/country’. Country codes: JPN, Japan; USA, United States of America; IRQ, Iraq; KOR, Korea.
DISCUSSION
NoVs are a crucial cause of viral gastroenteritis-related hospitalization [Reference Koopmans4]. Clarity on the virus and its pathogenesis and epidemiology is affected by the lack of in vitro cell-culture systems and in vivo small-animal models for human NoVs. In-depth information on this virus is essential for reducing the burden of NoV illness and hospitalization costs [Reference Lee and Pang9]. The molecular epidemiology of emerging NoV strains is of particular interest to researchers because NoV recombination, variants, and mutations occur frequently [Reference Dingle35, Reference Bull, Tanaka and White36]. Furthermore, uncommon and minor types of NoVs, which has the potential to dominate, have been consistently detected. In this study, NoV was detected in 64 out of 195 clinical samples from Korea, and positive samples were classified by genotyping using BLAST. The NoV genotypes GI.4 (n = 6), GI.6 (n = 2), GII.2 (n = 1), GII.3 (n = 2), GII.4 (n = 43), GII.6 (n = 3), GII.16 (n = 1), GII.17 (n = 5), and GII.21 (n = 1) were identified. Although GII.21 may have been consistently detected in many countries, it has not been detected as a dominant cause of gastroenteritis in certain countries [Reference Vega14, Reference Mans16–Reference Kim26].
The data generated from the current full-genome sequencing are expected to be helpful in the disease surveillance of sporadic gastroenteritis caused by non-dominant strains, such as NoV GII.21. BLAST results of the full-genome sequence of strain JW confirmed it to be a GII.21-type NoV, and ORF1, ORF2, and ORF3 genes showed very high similarity with other NoV GII.21 strains. Phylogenetic analysis based on the full-length genomic sequence and ORF1, ORF2, and ORF3 sequences showed the highest identity with NoV GII.21 (strain YO284). The amino acid sequences of ORF1, ORF2, and ORF3 also indicated the highest identity with strain YO284 (Fig. 1a – d ). However, alignment analysis of amino acid sequences of the VP1 region showed 21 sequence substitutions; of these, 14 amino acids were included in the P2 subdomain. In addition, alignment analysis of amino acid sequences of the five former strains and the recent strains indicated that most substitutions were present within the P2 subdomain (Fig. 3). The P2 subdomain is the highly variable part and the most exposed region of the structure [Reference Prasad31]. This suggests the possible formation of different epitopes or different protein structures because of a high rate of substitution in the P2 subdomain even among strains with high identity.
NoV GII.21 strains identified in Bangladesh were divided into three clusters based on the partial sequence of the S domain of the VP1 gene, which determines NoV genotype [Reference Nahar30]. Strain JW was included in GII.21.b1, and each cluster showed different amino acid substitution patterns (Fig. 2a , b ).
The P domain of VP1 interacts with HBGA in a strain-specific manner. GII.21 is divided into unique lineages on the basis of the binding interface with HBGAs, which is distinct from the conventional GII-binding interface [Reference Liu34]. Strain JW and three other GII.21 lineage strains had very high amino acid sequence similarities in B, N, and T loops, and the residues appeared to be well conserved. However, two amino acid sites were found to be substituted (Fig. 4). Such amino acid substitutions resulting from NoV point mutations occur frequently [Reference Dingle35].
Eight strains were isolated from the Hong Kong group in 2014–2015 (GenBank accession numbers KR921935, KR921936, KR921937, KR921938, KR921939, KR921940, KR921941, KR921942), and their alignment was analyzed with the full sequence of the ORF2 region. The strains were isolated around the same time as strain JW. The alignment of the amino acid sequence of the ORF2 region shows 100% identity with that of strain JW, and only one strain (GenBank accession number KR921940) showed one amino acid difference (data not shown). However, at the nucleotide level, only 2 nt differed between strain JW and strain CUHK-NS-626 (GenBank accession number KR921942). The third nucleotide of the codon was altered; however, this did not affect the encoded amino acid. NoV GII.21 isolated around the same time in Korea and Hong Kong exhibited high capsid protein sequence identity. In Korea, 10 strains were isolated from stream water in 2015 (GenBank accession numbers AMO28393, AMO28394, AMO28395, AMO28396, AMO28397, AMO28398, AMO28388, AMO28390, AMO28399, and AMO28400); these strains were isolated around the same time as strain JW. Amino acid sequences of the ORF2 region of the strains were aligned; the results showed high sequence identity (⩾98%) (data not shown). This indicates the epidemic potential of GII.21-type NoV.
Recently, a novel NoV recombinant strain, GII.4/GII.21, was isolated in Bangladesh [Reference Nahar30]. Furthermore, a variant of genotype GII.17 has been reported to be more predominant than GII.4 for acute gastroenteritis outbreaks in several countries [Reference de Graaf37–Reference Chan46]. GII.17 is genetically the closest to GII.21 [Reference Liu34], indicating that GII.21 also has the potential to dominate over GII.4. These studies highlight the importance of molecular epidemiology studies bolstered by whole-genome analysis to characterize non-dominant strains in addition to prevalent strains, and identify and track sporadic cases of NoV gastroenteritis globally.
To our knowledge, this is the first report of the full-genome sequence analysis of NoV GII.21 from Korea. This is a valuable contribution to the databases that enable viral evolutionary studies and molecular epidemiology studies. Furthermore, the information generated might facilitate the development of diagnostic tools and effective vaccines.
ACKNOWLEDGEMENTS
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C1781)), as well as by the Korea Ministry of Environment (MOE) as a Public Technology Program based on Environmental Policy (grant number: 2016000210002).
DECLARATION OF INTEREST
None.





