


INTRODUCTION
The order Chiroptera is considered the second largest group of mammals in the world, composed by ~20% of mammals with more than 1200 species that are present in all continents, except Antarctica [Reference Schipper1]. These animals are known to be hosts of important zoonotic virus (e.g. lyssavirus and hantavirus) and bacteria (e.g. Leptospira spp. and Pasteurella spp.) [Reference Mühldorfer2].
The genus Bartonella spp., belonging to the order Rhizobiales, comprises Gram-negative facultative intracellular α-proteobacteria that parasite mammals’ erythrocytes and endothelial cells [Reference Morse3]. These re-emerging agents have been identified in a wide variety of mammals and incriminated as important cause of diseases in humans and animals, causing different clinical manifestations ranging from self-limiting to potentially fatal syndromes [Reference Kosoy4–Reference Vieira-Damiani6].
Hemotropic mycoplasmas (hemoplasmas) are Gram-negative bacteria belonging to the order Mycoplasmatales, family Mycoplasmataceae, genus Mycoplasma [Reference Euzéby7]. These pathogens are known to cause manifestations ranging from asymptomatic and chronic disease to severe hemolytic anemia due to the capacity of adhesion to the erythrocyte surface, causing indentation or deformation of the target-cell membrane. Acute infected animals can also present anorexia, fever, icterus, and hypoglycemia, depending on the species involved [Reference Neimark8–Reference Tasker10]. These agents have also been detected in different mammals’ species, including humans [Reference dos Santos11–Reference Millán13].
Bartonella species can be transmitted by arthropod vectors, scratch, or by direct contact with blood or bodily fluids from animals [Reference Breitschwerdt14]. Similarly, hemoplasmas are supposed to be transmitted mainly by blood-sucking arthropod transmission or aggressive interactions between animals [Reference Neimark8, Reference Biondo9].
Bartonella spp. and Mycoplasma spp. have been detected in a variety of wild animals all over the world. In Brazil, these agents have been reported in several wild mammals, such as wild carnivores [Reference Guimarães15–Reference Fleischman17], deer [Reference Grazziotin18], peccary [Reference Castro19], non-human primates [Reference Santos20, Reference Bonato21], and rodents [Reference Vieira22–Reference Gonçalves26].
The presence of Bartonella spp. in Chiroptera has been reported in bats sampled in the UK [Reference Concannon27], Kenya [Reference Kosoy4], Taiwan [Reference Lin28], Peru [Reference Bai29], Nigeria [Reference Kamani30], Puerto Rico [Reference Olival31], Finland [Reference Lilley, Veikkolainen and Pulliainen32], Madagascar [Reference Brook33], Costa Rica [Reference Judson, Frank and Hadly34], Guatemala [Reference Bai35, Reference Wray36], French Guiana [Reference Davoust37], Gana [Reference Mannerings38], Algeria [Reference Leulmi39], and South Africa [Reference Dietrich40]. On the other hand, Mycoplasma spp. have already been molecularly detected in bats in the USA [Reference Millán13] and Spain [Reference Breitschwerdt14].
The present work aimed to investigate the occurrence and assessing the phylogenetic positioning of Bartonella spp. and Mycoplasma spp. in bats sampled from Brazil.
MATERIAL AND METHODS
Between December 2015 and April 2016, tissue and/or blood samples were collected from 162 bats, comprising 19 species belonging to four different families, namely Vespertillionidae, Phyllostomidae, Mollossidae, and Natalidae, sampled in five states in Brazil (Mato Grosso, Pará, Paraná, São Paulo, and Tocantins) (Fig. 1).

Fig. 1. Locations where bats were sampled in the present study. Each state is outlined in the map and the number of sampled animals is represented by a diamond symbol.
The bat captures were performed in accordance with the licenses obtained from the Brazilian Government Institute for Wildlife and Natural Resources Care (IBAMA) (license numbers 48306–1 and 10080–2), and were endorsed by the Ethics Committee of FCAV/UNESP University (Faculdade de Ciências Agrárias e Veterinárias, Universidade Estadual Paulista ‘Júlio de Mesquita Filho’, Câmpus Jaboticabal) n° 8189/15. The captures were performed as previously described [Reference Kunz, Kurta and Kunz41, Reference Peracchi and Nogueira42] using mist net (Zootech™, Curitiba, Paraná, Brazil) with variable sizes (3 m × 6 m, 3 m × 9 m, 3 m × 12 m). The nets were placed before sunset and kept closed until nightfall. Once opened, they were checked every 30 min until reaching sample effort of eight animals or until midnight. The animals were carefully removed from the mist nets with leather gloves, taken to the field laboratory to weight, determine species and gender, and then taken to the university's laboratory for euthanasia, necropsy procedures, and tissue samples collections.
DNA was extracted from 10 mg of spleen tissue and 25 mg of liver and heart tissues using the DNeasy™ Blood & Tissue Kit (Qiagen™, Valencia, California, USA), according to the manufacturer's instructions. The DNA quality was evaluated by concentration and 260/280 and 260/230 nm absorbance ratios using a spectrophotometer (Nanodrop, Thermo Scientific, Wilmington, Delaware, USA), in which the purity for DNA samples is considered when ratios are ~1·8 and ~2·0, respectively. Also, a conventional PCR (cPCR) assay, based on a 400 pb fragment of GAPDH gene [Reference Birkenheuer, Levy and Breitschwerdt43], was performed in order to evaluate the absence of inhibitors in DNA-extracted samples. Positive samples in the above-mentioned cPCR assay were submitted to additional Bartonella spp. and hemoplasmas PCR assays. All the cPCR assays were performed in a T100™ Thermal Cycler (BioRad™, Hercules, California, USA).
A previously described quantitative PCR (qPCR) protocol based on nuoG gene [Reference Bustin45] was used aiming to detect and quantify Bartonella spp. DNA copies (number of copies/μl) in bats’ biological samples. The qPCR assays were performed in 10 µl final volume reaction mixtures, containing 1 µl of DNA sample, 1·2 µM of each primer F-Bart (5′ – CAATCTTCTTTTGCTTCACC – 3′), R-Bart (5′ – TCAGGGCTTTATGTGAATAC – 3′), and hydrolysis probe TexasRed-5′ – TTYGTCATTTGAACACG-3′(BHQ2a-Q) – 3′, Master Mix 2× buffer (GoTaq™ Probe qPCR Master Mix, Promega Corporation, Madison, Wisconsin, USA) and ultra-pure sterilized water (Nuclease-Free Water, Promega Corporation, Madison, Wisconsin, USA) q.s.p. 10 µl. The amplification conditions were 95 °C for 3 min followed by 40 cycles at 95 °C for 10 s and 52·8 °C for 30 s [Reference André44]. PCR amplifications were conducted in low-profile multiplate unskirted PCR plates (BioRad™, Hercules, California, USA), using a CFX96 Thermal Cycler (BioRad™, Hercules, California, USA). Standard curves were constructed with serial dilutions of plasmid DNA (pIDTSMART – Integrated DNA Technologies, Coralville, Iowa, USA) (2·0 × 107–2·0 × 100 copies/μl), which encoded an 83 bp Bartonella henselae-nuoG gene fragment. The number of plasmid copies was determined in accordance following the formula:
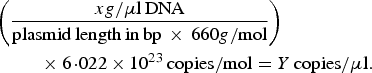 $$\eqalign{&\left( {\displaystyle{{xg/{\rm \mu l}\,{\rm DNA}} \over {{\rm plasmid}\,{\rm length}\,{\rm in}\,{\rm bp}\, \times \,660g/{\rm mol}}}} \right) \cr&\hskip2pc \times 6\!\cdot\!022 \times 10^{23} \,{\rm copies/mol} = Y\,{\rm copies/}{\rm \mu l}{\rm.}}$$
$$\eqalign{&\left( {\displaystyle{{xg/{\rm \mu l}\,{\rm DNA}} \over {{\rm plasmid}\,{\rm length}\,{\rm in}\,{\rm bp}\, \times \,660g/{\rm mol}}}} \right) \cr&\hskip2pc \times 6\!\cdot\!022 \times 10^{23} \,{\rm copies/mol} = Y\,{\rm copies/}{\rm \mu l}{\rm.}}$$
Each qPCR assay was performed including duplicates of each bat DNA sample and plasmids. All duplicates showing Cq values difference higher than 0·5 were retested in triplicates. Amplification efficiency (E) was calculated from the slope of the standard curve in each run using the following formula (E = 10–1/slope). The standard curves generated by 10-fold dilutions were used to determine the amount of DNA that could be detected with 95% of sensitivity [Reference Bustin45].
In order to perform the molecular characterization of Bartonella spp., DNA samples from positive bats in qPCR reactions were submitted to previously described cPCR assays targeting eight different genes, namely nuoG (400 bp) [Reference Colborn46], ribC (420 bp) [Reference Johnson47], gltA (750 bp) [Reference Norman48], rpoB (800 bp) [Reference Paziewska49], the intergenic region 16S–23SrRNA ITS (453–717 bp) [Reference Maggi and Breitschwerdt50], groEL (752 bp) [Reference Paziewska49, Reference Zeaiter51], fstZ (600 bp) [Reference Paziewska49], 16SrRNA (400 bp) [Reference Paziewska49], and pap-31 (564 bp) [Reference Maggi and Breitschwerdt52]. B. henselae DNA obtained from a naturally infected cat [Reference Bustin45] and sterilized ultrapure water (Nuclease-Free Water, Promega™, Madison, Wisconsin, USA) were used as positive and negative controls, respectively.
In order to amplify Mycoplasma spp. DNA, two cPCR assays based on 16SrRNA gene were performed, using two sets of primers, namely HemMycop16S-41s (5′-GYATGCMTAAYACATGCAAGTCGARCG-3′) and HemMyco16S-938as (5′ – CTCCACCACTTGTTCAGGTCCCCGTC – 3′) (fragment of ~800 bp), and HemMycop16S-322s (5′ – GCCCATATTCCTACGGGAAGCAGCAGT – 3′) and HemMycop16S-1420 as (5′ – GTTTGACGGGCGGTGTGTACAAGACC – 3′) (fragment of ~800 bp) [Reference Maggi53]. Five microliters of DNA were used as a template in 25 µl reaction mixtures containing 10× PCR buffer, 1·0 mM MgCl2, 0·8 mM deoxynucleotide triphosphate mixture, 1·5 U Taq Platinum DNA Polymerase (Life Technologies™, Carlsbad, California, USA), and 0·3 µM of each primer. Mycoplasma haemofelis DNA obtained from a naturally infected cat [Reference Miceli54] and ultra-pure sterile water (Nuclease-Free Water, Promega™, Madison, Wisconsin, USA) were used as positive and negative controls, respectively. PCR amplifications were performed at 94 °C for 2 min followed by 55 repetitive cycles of 94 °C for 15 s, 68 °C for 15 s, and 72 °C for 18 s, followed by a final extension at 72 °C for 1 min. The 16S rRNA-Mycoplasma spp. positive samples were additionally submitted to an RNaseP gene-Mycoplasma sp. (165 bp) cPCR assay using the oligonucleotides HemoMycoRNaseP30s (5′-GATKGTGYGAGYATATAAAAAATAAARCTCRAC – 3′) and HemoMyco RNaseP200as (5′ – GMGGRGTTTACCGCGTTTCAC – 3′). The conditions of amplification were the same as described above, except for the annealing temperature (59 °C) [Reference Maggi53].
The products obtained in all the cPCR assays were separated by electrophoresis on a 1% agarose gel stained with ethidium bromide (Life Technologies™, Carlsbad, California, USA) under 100 V/150 mA for 50 min. The gels were imaged under ultraviolet light (ChemiDoc MP Imaging System, Bio Rad™, Hercules, California, USA) using the Image Lab Software version 4.1.
Amplified products were purified using the Silica Bead DNA gel extraction kit (Thermo Fisher Scientific™, Waltham, Massachusetts, USA) and submitted to sequencing, which was performed using the BigDye™ Terminator v3·1Cycle Sequencing Kit (Thermo Fisher Scientific™) and ABI PRISM 310DNA Analyzer (Applied Biosystems™, Foster City, California, USA) [Reference Sanger, Nicklen and Coulson55].
The sequences obtained from positive samples were first submitted to a screening test using Phred-Phrap software version 23 [Reference Ewing and Green56, Reference Ewing57] to evaluate the electropherogram quality and to obtain consensus sequences from the alignment of the sense and antisense sequences. The BLAST program [Reference Altschul58] was used to analyze the sequences of nucleotides (BLASTn), aiming to browse and compare with sequences from an international database (GenBank) [Reference Benson59]. The consensus sequences obtained in the present study and those retrieved from GenBank were aligned using the Clustal/W software [Reference Thompson, Higgins and Gibson60] via Bioedit version 7.0.5.3 [Reference Hall61]. Phylogenetic inference was based on Bayesian Inference (BI) and Maximum Likelihood (ML) methods. The BI analysis was performed with MrBayes 3·1·2 [Reference Ronquist and Huelsenbeck62] via CIPRES Science Gateway [Reference Miller, Pfeiffer and Schwartz63]. Markov Chain Monte Carlo (MCMC) simulations were run for 106 generations with a sampling frequency of every 100 generations and a burn-in of 25%. The ML analysis was inferred with the W-IQ-Tree tool available online (http://iqtree.cibiv.univie.ac.at/) [Reference Trifinopoulos64, Reference Nguyen65] using 1000 bootstrapping replicates. The best model of evolution was selected by the program jModelTest2 (version 2.1.6) on XSEDE [Reference Darriba66], under the Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC) [Reference Posada and Buckley67]. All the trees were examined in Treegraph 2·0·56–381 β [Reference Stover and Muller68].
RESULTS
A total of 122 liver tissue samples, 107 spleen tissue samples, 56 heart tissue samples, and 40 blood samples collected from 162 bats in different states of Brazil were used in this study. So, out of 325 bats’ biological samples, 322 samples were positive in cPCR for GAPDH gene (Table 1). The mean and standard deviation of the values of concentration and absorbance ratios (260/280 and 260/230 nm) are shown in Table 2.
Table 1. Number of bat species positive for Bartonella spp. and hemoplasmas in Brazil, according to locality and tissue sampled

Table 2. Mean and standard deviation values of concentration and absorbance ratios (260/280; 260/230 nm) of DNA samples extracted from each type of bats’ biological sample

Seventeen (5·28%) out of 322 samples were positive for Bartonella spp. in qPCR assays based on the nuoG gene, including four samples collected from three bats from the state of Paraná (Sturnira lilium (n = 3)); six from the state of Pará (Phyllostomus hastatus (n = 1), Carollia perspicillata (n = 4), and Natalus espiritosantensis (n = 1)); and seven from the state of Tocantins (C. perspicillata (n = 2) and Glossophaga soricina (n = 5)). The efficiency, R 2, slope, and Y-intercept of reactions ranged from 90·8% to 98·6% (mean = 95·09%), 0·987 to 1 (mean = 0·995), −3·355 to −3·566 (mean = −3·447), and 37·284 to 38·871 (mean = 38·067), respectively. The quantification positive nuoG Bartonella spp. ranged from 4·4 × 103 to 6·95 × 103 copies/μl (Table 3).
Table 3. Positive samples for Bartonella sp. in qPCR assays based on nuoG gene with the reactions parameters

Thirteen (76·47%) out of 17 positive samples in the qPCR were also positive for at least one target gene in cPCR assays for Bartonella spp. Four (30·77%) of them were positive for the ftsZ gene, three (23·07%) for the nuoG gene, three (23·07%) for the ribC gene, two (15·38%) for the groEL gene, and one (7·69%) was positive for the rpoB gene. None of them were positive for pap-31 and 16SrRNA genes and for the intergenic region 16S–23S rRNA (ITS). Due to the low intensity of some amplified products, which precluded high-quality sequencing, only seven Bartonella spp. sequences were obtained (nuoG (n = 3), gltA (n = 2), rpoB (n = 1), ftsZ (n = 1)). The sequences obtained were deposited to the GenBank under accession numbers KY356752–KY356758.
Regarding the occurrence of hemoplasmas among bats, while 45 samples (13·97%) were positive for the first 16S rRNA protocol (using HemMycop16S-41s and HemMyco16S-938 as primers), 14 samples (4·34%) were positive for the second 16S rRNA protocol (using HemMycop16S-322s and HemMycop16S-1420 as primers). Among positive animals, 33 samples were collected from 18 bats in the state of Paraná (Molossus molossus (n = 10), S. lilium (n = 8), Eptesicus spp. (n = 1)); seven samples were collected from bats in the state of Pará (Artibeus planirostris (n = 3), M. molossus (n = 2), Eumops auripendulos (n = 1), Myotis nigricans (n = 1)); four from bats in the state of Tocantins (G. soricina (n = 4)); and one from a bat in the state of Mato Grosso (Molossus rufus). Twelve (26·66%) out of 45 samples positive for hemoplasmas based on 16S rRNA gene were also positive at RNAaseP gene-cPCR assay. A total of five (1·55%) samples were positive for the three performed protocols. Unfortunately, only five 16S rRNA hemoplasmas sequences (Paraná (n = 4), Pará (n = 1)), detected in specimens of M. molossus, were obtained due to the low intensity of some amplified products, which precluded high-quality sequencing. The sequences were deposited to the GenBank data under the accession numbers KY356747–KY356751. Only one bat sampled in Tocantins state belonging to G. soricina species was positive for both Bartonella sp. and Mycoplasma sp.
Based on BLAST analysis, the found Bartonella nuoG sequences (n = 3) showed 89–90% identity to B. taylorii (GenBank accession number EF659943) and 92% to B. koehlerae (GenBank accession number EF659942). Two Bartonella gltA sequences showed 98–100% identity to Bartonella sp. from bats sampled in Costa Rica (GenBank accession numbers KJ816674 and KJ816690). One Bartonella rpoB sequence showed 89% identity to Bartonella sp. Khabarovsk from Asian mammals (GenBank accession number AB779537). Finally, one Bartonella ftsZ sequence showed 98% identity to Bartonella sp. Honshu from deer sampled in Japan (GenBank accession number AB703117). The query coverage ranged from 96% to 100% in all BLAST analyses run for Bartonella sequences. In the phylogenetic inferences based on ML and BI methods (Figs 2–5), the Bartonella sequences obtained in the present study were mainly closely related to other Bartonella sequences obtained from New World bats. When present, Bartonella sequences obtained from Old World bats clustered separately with high branch support.

Fig. 2. Concatenated phylogenetic analysis of Bartonella nuoG and gltA sequences (3370 bp after alignment) based on the topology generate on the BI method. The values of support of posterior probability/bootstrap higher than 50% are shown in each branch. The sequences of the present work were highlighted in red composing two different clusters. One comprising three sequences of the present study (red branches) clustering with sequences obtained from bats from Guatemala and Peru and from rodents sampled in Brazil (sequences highlighted in blue) and in the USA with a 59/71% of probability. The other cluster (green branches) comprises one sequence obtained in the present study with sequences obtained from bats sampled in Costa Rica, Peru, and Guatemala, with 66% of probability. Brucella abortus and Ochrobactrum anthropi were used as outgroups.

Fig. 3. Phylogenetic analysis of Bartonella rpoB sequences (1500 bp after alignment) based on the topology generate on the BI method. The values of support of posterior probability/bootstrap higher than 50% are shown in each branch. The sequence of the present work is highlighted in red. The sequence was positioned alone in one branch and clustered with Bartonella sp. from wild mammals from Asia with 94/85% of probability (red branches). Brucella abortus and Ochrobactrum anthropi were used as outgroups.

Fig. 4. Phylogenetic analysis of Bartonella ftsZ sequences (790 bp after alignment) based on the topology generate on the BI method. The values of support of posterior probability/bootstrap higher than 50% are shown in each branch. The Bartonella sequence of the present work was highlighted in red. The Bartonella sequences highlighted in blue were previously detected in rodents from the same research group. The Bartonella sequence detected in a specimen of Carollia perspicillata was positioned alone in one branch and clustered with Bartonella sp. detected in a deer from Asia and other Bartonella spp. with 100% of probability (red branches). A Bartonella genotype detected in a bat sampled in Finland was closely positioned with the sequence of the present study, with 100/75% of probability (green branches). Brucella abortus and Ochrobactrum anthropi were used as outgroups.

Fig. 5. Phylogenetic analysis of hemoplasmas 16SrRNA sequences (1770 bp after alignment) based on the topology generate on the BI method. The values of support of posterior probability/bootstrap higher than 50% are shown in each branch. The sequences of the present work were highlighted in red and placed together in a same branch with 100/95% of probability (red branches). Mycoplasma pneumoniae was used as outgroup.
The concatenated phylogenetic assessment of positive sequences for both nuoG (400 pb) and gltA (750 pb) genes showed a very similar topology in BI and ML methods, forming two different clusters. The sequences from bats detected in a specimen of S. lilium sampled in the state of Paraná was closely related to Bartonella sequences detected in a Hippoboscidae bat fly (Aspidoptera delatorrei) from Costa Rica and in a specimen of C. perspicillata from Guatemala with 100/100% of branch support. A Bartonella genotype detected in a specimen of C. perspicillata sampled in Tocantins was positioned in the same cluster (with 59% and 71% of support in BI and ML analyses, respectively) than genotypes detected in bats from Peru and Guatemala, rodents from Brazil and the USA, and from an ectoparasite (Polygenis gwyni) collected from rodents also in the USA. Moreover, a Bartonella sequence obtained in a specimen of G. soricina sampled in Tocantins state was positioned in a more basal clade and closely related to Bartonella genotypes detected in specimens of Carollia sowelli and its Streblidae dipteran Strebla guajiro from Costa Rica (with branch support of 100%) and genotypes detected in bats from Peru and Guatemala with 66% in both analyses (ML and BI) (Fig. 2).
The Bartonella rpoB sequence obtained in a specimen of S. lilium sampled in the state of Paraná was positioned alone in a branch, but closely related to two Bartonella genotypes detected in wild rodents from Asia (AB779537; AB290276) and B. taylorii (AF165995), with a branch support of 85% in ML and 94% of probability in BI analyses (Fig. 3).
The Bartonella ftsZ sequence obtained in a specimen of C. perspicillata sampled in the state of Pará was positioned alone in a branch, but closely related (100% of branch support) to a Bartonella genotype detected in a deer in Japan (AB703117), Bartonella chomelii (KM215689/AB290193) and Bartonella schoenbuchensis (AF467765) (Fig. 4).
All five 16S rRNA hemoplasmas sequences detected in specimens of M. molossus sampled in the states of Paraná and Pará shared identity (93–96%) with Mycoplasma coccoides (AY171918), with query coverage ranging from 87% to 100%. Additionally, the found hemoplasma sequences showed 92–96% identity with an uncultured Mycoplasma sp. detected in an Akodon rodent from Brazil (KT215636), with query coverage ranging from 87% to 99%. All the hemoplasmas sequences obtained in the present study clustered together as a monophyletic group with branch support of 95/100% by ML and BI analyses. This group of sequences was positioned between M. coccoides (AY171918) and hemoplasma genotypes detected in rodents from Brazil (KM203857; KT215636). The hemoplasmas sequences detected in bats in the present study were positioned in the Haemofelis group (Figure 5).
DISCUSSION
The present work reported the occurrence and molecular characterization of Bartonella spp. and Mycoplasma spp. among bat's population sampled in five different states of Brazil. This group of mammals has been incriminated as potential reservoirs for several pathogens due to their high mobility, worldwide distribution, and social behavior [Reference Bai35].
The Bartonella occurrence found in the present study (5·28%) was similar to that found in bats (3·38% (13/384)) sampled in Swaziland and South Africa, Africa [Reference Dietrich40]. Overall there is a relatively higher occurrence of Bartonella in bats sampled in the Old World, such as in Algeria (60% (6/10) by qPCR) [Reference Leulmi39], Nigeria (51·35% (76/148) by qPCR and 15·54% (23/148) by culture) [Reference Kamani30], Kenya (32·02% (106/331) by culture) [Reference Kosoy4], and Madagascar (44·68% (21/47) by PCR) [Reference Brook33]. In neotropical species, Bartonella species have been already detected by solid culture followed by cPCR or only by PCR in bats sampled from Guatemala, Peru, and Costa Rica, whose overall prevalence ranged from 24·1% to 33·3% [Reference Bai29, Reference Judson, Frank and Hadly34, Reference Bai35].
Although a low occurrence of Bartonella in S. lilium and G. soricina bat specimens is reported [Reference Bai35], these animals together with C. perspicillata were the main Chiroptera species that showed positivity for Bartonella in the present study. Despite the high number of Molossus spp. individuals (29·01% (47/162)) sampled in the present study, none of them was positive for Bartonella sp., as previously reported [Reference Bai29] as well. Bartonella genotypes have already been described previously in the bat species sampled in the present work [Reference Bai29, Reference Bai35], except for N. espiritosantensis.
Even though it is suggested that the spleen is the sample of choice to detect Bartonella spp. [Reference Gutiérrez69], no difference was observed among the tissues collected in the present study, since seven were from blood, six from splenic tissue, and four from liver tissue.
Bartonella host transitions are supposed to occur with bats within the same family, but transitions between bats belonging to different families, superfamilies and suborders seem to occur infrequently [Reference Mckee70]. It had already been hypothesized that a different pressure on the coevolutionary relationships between Old and New World bats host species, since Old World species seem to be more rustic and show a longer evolutionary time for the establishment of this sort of relationship [Reference Lei and Olival71]. This hypothesis could explain the different positioning of Bartonella genotypes detected in neotropical bats, including those sampled in the present study, when compared with those from Africa (Megachiroptera/Yangpteroptera). However, it is noteworthy to observe that Bartonella genotypes detected in bats from the present study were closely related to those detected in wild mammals, such as rodents and deer from Asia, based on ftsZ and rpoB phylogenetic analyses. Regarding to these findings, intra- and inter-specific Bartonella transmission has been already suggested, since different Bartonella species/genotypes can be found in only one individual [Reference Bai35].
Interestingly, Bartonella genotypes detected in Brazilian rodents were positioned in the same clade with the sequences obtained from bats sampled in the present study with a 59/71% of node support based on the concatenated nuoG and gltA analysis. However, a Bartonella genotype detected in a specimen of G. soricina sampled in the state of Tocantins was positioned distant from the other Bartonella genotype detected in the state of Paraná, clustering with a Bartonella genotype detected in specimens of C. sowelli and its Streblidae dipteran, namely S. guajiro from Costa Rica, based in gltA/nuoG concatenated analysis. These findings suggest the occurrence of different Bartonella genotypes among bats in Brazil. On the other hand, Bartonella genotypes amplified from different tissue samples (spleen and liver) belonging to a specimen of S. lilium sampled in Paraná state clustered together, probably representing the same Bartonella genotype.
Regarding the occurrence of hemoplasmas among bats, Mycoplasma spp. has been poorly reported in this group of mammals. Despite the Molossus spp. was the main group of bats positive for hemoplasmas in the present study, to the best authors’ knowledge, this is the first molecular detection of hemoplasmas in eight bat species, namely A. planirostris, Eptesicus sp., Eumops auripendulus, G. soricina, M. molossus, M. rufus, M. nigricans and S. lilium.
High prevalence rates for hemoplasmas have been found in small populations of bats sampled in the USA (47% (32/68)) [Reference Mascarelli12] and Spain (41·93% (13/31)) [Reference Millán13]. Interestingly, hemoplasmas genotypes detected in bats from both studies were similar to each other, but different for Mycoplasma genotypes detected in bats sampled in Brazil. The hemoplasmas sequences detected in bats sampled in the USA and Spain clustered with ‘Candidatus Mycoplasma haemohominis’ that was detected in a human patient in England [Reference Steer72], and with Mycoplasma haemomuris, which has been detected in domestic, laboratory, and wild rodents [Reference Neimark8]. Based on the phylogenetic positioning, hemoplasmas genotypes detected in bats in the present study seem to be different from those previously detected in bats sampled in the USA and Spain, since comprised a monophyletic group with high node support and closely related to M. coccoides.
Despite the higher positivity to hemoplasmas than to Bartonella spp., only one animal was co-positive for both studied pathogens. The pathogenic potential of both group of pathogens in bats have not been investigated.
Finally, it is important to highlight that Bartonella and Mycoplasma genotypes detected in bats from the present study were closely related to previously reported genotypes detected in rodents. From an evolutionary point of view, bats and rodents are large groups belonging to ancient orders of mammals [Reference Lei and Olival71], which have shown a high adaptation as hosts for different groups of pathogens.
To the best of authors’ knowledge, the present work presented the first molecular evidence of circulation of Bartonella and hemoplasmas among bats in Brazil. Future studies aiming at assessing the role of bats as reservoirs for Bartonella and Mycoplasma species showing zoonotic potential are much needed.
ACKNOWLEDGEMENTS
The authors are thankful to all the staff and trainees of the Infectious and Parasitic Diseases Laboratory, Mammals Biodiversity of Southern Brazil Laboratory and Wildlife Service (SAAS) of Universidade Estadual do Centro-Oeste (UNICENTRO) for the support given during the animal's captures in the state of Paraná.
This work was supported by ‘Fundação de Amparo à Pesquisa do Estado de São Paulo’ (FAPESP) for the financial support (#2015/14896–1) and P. Ikeda Msc. scholarship (#2015/04773–0).
DECLARATION OF INTEREST
None.
ETHICAL STANDARDS
The authors assert that all procedures contributing to this work comply with the ethical standards according to the Ethical Principles in Animal Research adopted by the Brazilian College of Animal Experimentation, and to the 2000 Report of the AVMA Panel on Euthanasia (American Veterinary Medical Association, 2001).








