


Coronaviruses (CoVs) are a group of enveloped, single-stranded RNA, positive-strand viruses with a corona-like morphology. CoVs have diverse animal hosts ranging from mammalian to avian species, causing mainly enteric and respiratory diseases of varying severity.
In particular, the severe acute respiratory syndrome-associated coronavirus (SARS-CoV), is the aetiological agent of SARS disease, which caused a global human epidemic in 2002–2003, resulting in more than 8000 infections and more than 700 deaths [Reference Peiris, Guan and Yuen1].
Several studies indicate that bats might be the natural reservoir for several viruses closely related genetically to SARS-CoV, the SARS-like coronaviruses (SARS-like CoVs), and that all mammalian CoVs were derived from ancestor viruses residing in bats [Reference Woo2]. The association between CoVs and bats is a worldwide phenomenon, in fact, after the initial discovery of this virus in Chinese bats [Reference Lau3, Reference Poon4], recent studies have confirmed their presence in bats from Europe, Africa, and South and North America.
In particular, bats belonging to the genus Rhinolophus (suborder Microchiroptera, family Rhinolophidae) are reservoir hosts for several species of CoVs, including the SARS-like CoVs [Reference Lau5–Reference Rihtaric7].
The recent discovery of novel CoVs in bats has led the Coronavirus Study Group of the International Committee for Taxonomy of Viruses (ICTV) to propose a reclassification of the family Coronaviridae into two subfamilies, Coronavirinae and Torovirinae. The subfamily Coronavirinae comprises only three genera, Alphacoronavirus, Betacoronavirus, and Gammacoronavirus, which have replaced the traditional antigenic groups 1, 2, and 3, respectively [Reference Woo2]. All mammalian CoVs, including all bat CoVs detected up to now and SARS-CoV, belong to the Alphacoronavirus and Betacoronavirus genera, whereas all avian CoVs belong to the genus Gammacoronavirus.
This study examined the presence of CoVs in Italian colonies of the greater horseshoe bat Rhinolophus ferrumequinum, which is one of about 70 species of the genus Rhinolophus widely distributed intropical and temperate regions of Asia, Africa, Europe, and Australia. All bat species in Italy are protected by national and international laws. R. ferrumequinum is a vulnerable species according to the Italian Mammals Red List 2007 because of human disturbance of its roosting sites, mainly caves and abandoned buildings, and of the progressive depletion and fragmentation of its habitats.
Fifty-two R. ferrumequinum specimens were captured from different roosts spread over peninsular Italy, including caves, mines and abandoned houses, over a 5-month period (from July to November 2009). All captures were authorized by the Italian Ministry of the Environment and were part of a Ph.D. project on R. ferrumequinum conservation.
Bats were caught using harp traps and put singly into cotton bags prior to subsequent investigation. Once species, sex, age category (juvenile, subadult, adult), forearm length and weight were determined, bats were released at their capture site.
Faeces (41 samples) or anal swabs (11 samples, collected if the bat did not produce a fresh bolus during handling) were collected and kept in 2 ml RNAlater RNA stabilization reagent (Qiagen, Germany) to allow the conservation of RNA during transport. Samples were then conserved at −80°C before processing.
Viral RNA extraction from faecal samples was performed by using the RNeasy Mini kit (Qiagen) according to the manufacturer's instructions and CoV screening was performed by reverse transcription–polymerase chain reaction (RT–PCR) using conserved primers for the RNA-dependent RNA polymerase gene (forward: 5′-GGTTGGGACTATCCTAAGTGTGA-3′ and reverse: 5′-CCATCATCAGATAGAATCATCATA-3′) frequently used for CoV detection and subsequent phylogenetic analysis [Reference Lau3, Reference Poon4, Reference Lau5, Reference Rihtaric7].
RT–PCR was performed using the Titanium One-Step RT–PCR kit (Clontech, USA), in a total volume of 50 μl, using a RNA extract of feline coronavirus (FCoV)-positive sample as positive control. The mixtures were amplified as follows: (i) initial reverse transcription at 50°C for 60 min, and 94°C for 5 min; (ii) 40 cycles of amplification with 1 cycle consisting of 1 min at 94°C, 1 min at 48°C, and 1 min at 68°C; (iii) final further elongation at 68°C for 10 min. Standard precautions were taken to avoid PCR contamination, and water controls included in all RT–PCR assays did not show false-positive results.
PCR products (5 μl each) were electrophoresed in 2·0% agarose gels in standard TAE buffer and visualized by UV light after staining with GelRed (Biotium, USA).
Positive PCR products of the expected size of 440 bp, were detected in 2/52 samples examined with 3·8% virus prevalence. Both positives were from faecal samples, whereas none of the 11 anal swab specimens tested positive by RT–PCR. It is possible that the absence of positive results in the anal swab specimens was due to the small quantity of faecal material collected with this technique.
The two positive samples, 771/09-5 and 893/09-11, came from different Italian regions, one in the north and the other in the south, from the provinces of Modena and Foggia, respectively.
Nucleotide sequence of the sample 893/09-11 was obtained by direct sequencing of PCR product with an ABI 3730xl 96-capillary DNA Analyzer (Applied Biosystems, USA); whereas for sample 771/09-5, it was not possible to obtain a reading sequence of good quality, probably due to the limited amount of genetic material in the faecal sample. The 893/09-11 sequence was aligned and compared to 48 selected human and animal CoV sequences available from the GenBank database using ClustalW software implemented in BioEdit version 7.0.5.
The phylogenetic relationships were evaluated using MEGA version 4.0; pairwise genetic distances were calculated by the Tamura 3-parameter model and phylogenetic trees were constructed using the neighbour-joining method. Bootstrap values were determined by 1000 replicates to assess the confidence level of each branch pattern (Fig. 1).
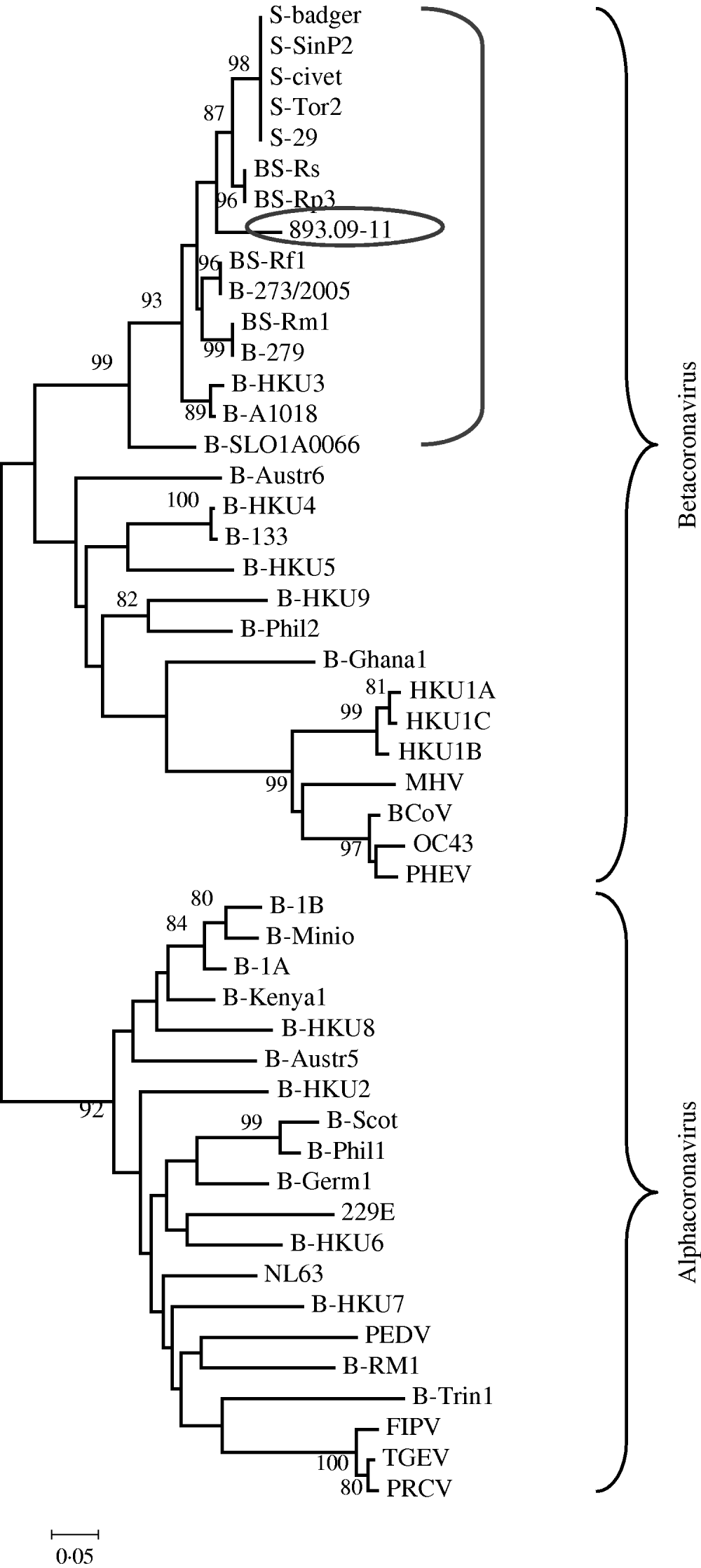
Fig. 1. Phylogenetic relationship of 893/09-11 with 48 alphacoronaviruses and betacoronaviruses obtained from GenBank. The tree was generated based on a fragment of RNA-dependent RNA polymerase region by the neighbour-joining method in the MEGA program. Numbers to the left of branches are bootstrap values ⩾80%, calculated from 1000 bootstrap replicates. Circled: 893/09-11; bracketed: coronavirus group 2b, SARS-related coronavirus and some unclassified coronavirus. Sequences obtained from GenBank: S-civet (AY304488), S-badger (AY545919), S-29 (CQ903025), S-SinP2 (AY559089), S-Tor2 (NC_004718), BS-Rs (NC_013664), BS-Rp3 (NC_009693), BS-Rf1 (NC_009695), B-273/2005 (DQ648856), BS-Rm1 (NC_009696), B-279 (DQ648857), B-HKU3 (NC_009694), B-A1018 (DQ648795), B-SLO1A0066 (GQ404795), B-Austr6 (EU834950), B-HKU4 (NC_009019), B-133 (NC_008315), B-HKU5 (NC-009020), B-HKU9 (NC_009021), B-Phil2 (AB539081), B-Ghana2 (FJ710053), HKU1A (NC_006577), HKU1B (AY884001), HKU1C (DQ339101), MHV (NC_001846), BCoV (NC_003045), OC43 (NC_005147), PHEV (NC_007732), B-1B (NC_010436), B-Mino (AY864197), B-1A (NC_010437), B-Kenya1 (GQ920838), B-HKU8 (NC_010438), B-Austr5 (EU834951), B-HKU2 (NC_009988), B-Scot (NC_009657), B-Phil1 (AB539080), B-Germ1 (EU375875), 229E (NC_002645), B-HKU6 (DQ249224), NL63 (NC_005831), B-HKU7 (DQ249226), PEDV (NC_003436), B-RM1 (EF544565), B-Trin1 (EU769558), FIPV (AY994055), TGEV (NC_002306), PRCV (DQ811787).
The comparison of RdRp gene fragment reference sequences to the sequence of sample 893/09-11 showed a percentage of nucleotide identity ranging from 86% to 91% with unclassified CoV group 2b, and a value of 89% of similarity with SARS-CoVs. The percentage of nucleotide identity decreased to values which varied from 69% to 67% with bat CoVs of the genus Betacoronavirus, to 57–64% with bat CoVs of the genus Alphacoronavirus and to 53–59% with mammalian non-bat CoVs of both genera, Alphacoronavirus and Betacoronavirus (with the exception of 64% of human CoV NL63). Furthermore, sample 893/09-11 had highly variable values of nucleotide identity when compared to some unclassified bat CoVs, e.g. with A1018/2005 and SLO1A0066/2008/SVN strains it had a percentage of 84–87%, while the nucleotide identity with other strains, such as Kenya bat CoV BtKY42 and Bat CoV Hipposideros/GhanaKwam/10/2008, was 64% (data not shown).
From these values of nucleotide identity it can be assumed that a strict correlation exists between 893/09-11 and CoVs related to SARS (human SARS-CoV, civet, and ferret badger SARS-CoV, and SARS-related Rhinolophus bat CoV) rather than to other bat and mammalian CoVs.
These findings are supported by the phylogenetic tree created with this fragment of the RdRp gene that suggests sample 893/09-11 was grouped together with SARS-CoVs, SARS-related CoVs and a few unclassified CoVs (Fig. 1). Data are supported by bootstrap values >90%.
To our knowledge, this is the first report of the detection of CoVs in bats of the genus Rhinolophus in Italy. The genetic analysis of the sequence obtained in this study has supported evidence that the virus is related to the SARS-like CoVs from the genus Betacoronavirus. This finding is similar to that obtained from another study conducted in Slovenia [Reference Rihtaric7].
In our study we detected a virus prevalence of 3·8%, which appears to be lower than those found in Asia or in other European countries, where it is usually higher than 10%, reaching 30–40% in particular bat species and geographical locations. This low number of infected bats may signify a genuine low prevalence of CoV infection in Italian bats. It may be that the climatic or geographical conditions influence the social behaviour of bats, reducing the length of displacements and meetings between bats, thus reducing virus transmission (or circulation).
Alternatively, the low number of infected bats detected may also be due to small amounts of viral RNA in the faecal samples or a suboptimal match of the primers used for RT–PCR with genetically more divergent CoVs.
In conclusion, our report provides some information about the ecology and evolution of CoVs in wildlife, confirming the widespread association between CoVs and several bat species in Europe, as recently reported in Germany, The Netherlands, and Slovenia [Reference Rihtaric7–Reference Reusken9].
SARS-like CoVs are widespread in Asia, Africa, and Europe, and are mainly associated, in Asia and Europe, with horseshoe bats. Bat species belonging to the genus Rhinolophus have a greater importance in CoV ecology compared to other bat species for the great variety in alphacoronaviruses and betacoronaviruses identified therein and, in particular, for the close correlation that some CoVs detected in Rhinolophus spp. (SARS-related Rhinolophus bat CoV) have with SARS-CoV. In the light of these results, it is likely that SARS-related Rhinolophus bat CoVs have a key role in the evolutionary history of SARS-CoV and in the possible future emergence of new zoonotic CoV variants [Reference Tang6, Reference Lau10].
The detection of CoVs in Italian bat populations and the close genetic correlation of Italian Rhinolophus CoV with human CoVs, underline the necessity to conduct further investigation to assess the prevalence of CoVs in Italian horseshoe bats and to determine the ecological relationships between bats, humans and other animals. Further understanding of virus maintenance in natural hosts, molecular mechanisms underlying interspecies jumping and zoonotic potential of Rhinolophus CoV are essential to contain emerging viruses and concurrently protect our wildlife heritage.
DECLARATION OF INTEREST
None.

