


The muscular dystrophies are a group of hereditary degenerative disorders associated with progressive muscle weakness. They can be transmitted as autosomal dominant, autosomal recessive, or X-linked traits; sporadic cases may also arise as a result of de novo mutation. Early presentation during childhood is generally associated with a more severe phenotype. Traditionally, the diagnosis is based on clinical and pathological features; more recently, the majority of muscular dystrophies have been classified based on molecular genetic confirmation.Reference Narayanaswami, Weiss, Selcen, David, Raynor and Carter 1 Research toward effective therapies is ongoing. Regional population-based prevalence estimates for the muscular dystrophies exist, but more precise pooled estimates representing the global burden of disease are unavailable. Robust pooled estimates are essential to facilitate the interpretation of clinical studies on molecular epidemiology, natural history, and impact of potential treatments. In addition, analysis of economic impact and health care burden are contingent upon precise prevalence estimates. We previously reported on the epidemiology of Duchenne and Becker muscular dystrophies.Reference Mah, Korngut, Dykeman, Day, Pringsheim and Jette 2 The purpose of this study is to systematically evaluate the worldwide incidence and prevalence of other muscular dystrophies including myotonic dystrophy, facioscapulohumeral dystrophy, limb girdle muscular dystrophy, and congenital muscular dystrophy.
Myotonic dystrophy is an autosomal dominant disorder associated with clinical myotonia, progressive muscular weakness, and extramuscular manifestations such as cardiac arrhythmia and endocrine dysfunction. It is classified based on molecular genetic testing showing an expansion of trinucleotide (CTG) repeats on chromosome 19q13.3 for type 1 and tetranucleotide (CCTG) repeats on chromosome 3q21.3 for type 2 disease.Reference Brook, McCurrach, Harley, Buckler, Church and Aburatani 3 , Reference Ranum, Rasmussen, Benzow, Koob and Day 4 Myotonic dystrophy type 1 can be subdivided into several clinical phenotypes depending on the age of presentation, including congenital, early childhood, adult, and late-onset forms. Congenital myotonic dystrophy type 1 is associated with multiple joint contractures, severe hypotonia, and generalized weakness; dysphagia and respiratory insufficiency leads to increased mortality during the neonatal period.Reference Campbell, Sherlock, Jacob and Blayney 5
Facioscapulohumeral dystrophy is an autosomal dominant disorder resulting from deletions within the D4Z4 repeat region located on chromosome 4q35 for type 1; mutations of SMCHD1 on chromosome 18p11.32 in association with a permission chromosome 4 allele account for the majority of type 2 disease.Reference Padberg, Lunt, Koch and Fardeau 6 , Reference Lemmers, Tawil, Petek, Balog, Block and Santen 7 It results in progressive atrophy and, frequently, asymmetrical weakness in a descending pattern. Early-onset facioscapulohumeral dystrophy is associated with more severe weakness as well as central nervous system involvement such as mental retardation, epilepsy, retinal vasculopathy, and sensorineural hearing loss.Reference Jardine, Koch, Lunt, Maynard, Bathke and Harper 8
Limb girdle muscular dystrophy refers to a heterogeneous group of autosomal muscular dystrophies with progressive weakness affecting predominantly the hip and shoulder girdles. It is further classified as either type 1 (dominant) or type 2 (recessive) disease based on the mode of inheritance, and labeled consecutively by letters of the alphabet according to the sequence of genes identified. It is related to mutations involving extracellular matrix or external membrane proteins, enzymes or proteins with putative enzymatic function, sarcolemma-associated proteins, nuclear membrane proteins, sarcomeric proteins, and other as-yet unspecified disorders.Reference Mercuri and Muntoni 9
Congenital muscular dystrophy refers to a heterogeneous group of early-onset muscular dystrophies. Affected children are usually symptomatic at birth or before their first 6 months of life. The salient features include hypotonia, muscle weakness, and reduced deep tendon reflexes, with or without joint contractures. Feeding and respiratory insufficiency are common; additional features may include microcephaly, eye anomalies, cerebral malformation, joint laxity, muscle atrophy, or hypertrophy. It is further subdivided into disorders involving: (1) the basal lamina or extracellular matrix proteins; (2) alpha- dystroglycanopathy; (3) sarcoplasmic reticulum calcium release channel; (4) endoplasmic reticulum proteins; (5) nuclear envelope proteins; (5) mitochondrial membrane proteins; and (6) other unspecified dystrophies.Reference Bönnemann, Wang, Quijano-Roy, Deconinck, Bertini and Ferreiro 10
This study was part of a larger initiative funded by the Public Health Agency of Canada to facilitate better understanding of the burden of neurological illnesses in Canada and worldwide.Reference Caesar-Chavannes and MacDonald 11
Methods
Search Strategy
The search strategy was developed by the study authors with expertise in neurology and epidemiology and in consultation with a research librarian with systematic review expertise. The comprehensive systematic review was conducted on January 28, 2011, using Medline and EMBASE databases to identify worldwide population-based studies on the incidence and prevalence of muscular dystrophies. References were exported and managed using EndNote X5.
Study Selection
Two independent reviewers screened abstracts to determine eligibility for full-text review. Abstracts and titles of all references were screened independently, in duplicate, to identify original research articles reporting on the prevalence or incidence of muscular dystrophies, including congenital muscular dystrophy, myotonic dystrophy, facioscapulohumeral muscular dystrophy, limb girdle muscular dystrophy, Emery-Dreifuss muscular dystrophy, oculopharyngeal muscular dystrophy, severe childhood autosomal recessive muscular dystrophy, Duchenne muscular dystrophy, or Becker muscular dystrophy. Articles were included if they met the following criteria: (1) original research, (2) population-based, (3) reported an incidence or prevalence estimate of any muscular dystrophy, and (4) published in English or French. Studies were considered population-based if they used a sampling method meant to be representative of the entire population and/or were completed in a defined geographic area of known population size. Studies were excluded if they were clearly not population-based, did not provide an estimate of incidence or prevalence, reported non-original data (i.e. reviews, letters, editorials), or if the study data were collected before 1985. The decision to include publications from 1985 onward was based on the advent of molecular genetic testing as well as magnetic resonance imaging studies after 1985. The reference lists of excluded non-original data studies and the studies that were included in the review were manually searched for additional articles.
Data Abstraction
Two reviewers independently screened the full-text articles of abstracts identified in the first phase. Demographic data including age characteristics, race, sex, and geographic location were recorded. Diagnostic data were also collected, as were the sources of those data and the definitions/diagnostic criteria for muscular dystrophy. Incidence and prevalence estimates of muscular dystrophy from each study were recorded, along with any stratification by age or gender, if provided. Agreement at the abstract review stage was calculated using the Kappa statistic (see http://www.cochrane.org/handbook/726-measuring-agreement). Disagreements between reviewers during abstract screening, full-text review, and/or data extraction were resolved by consensus and the use of a third reviewer as required.
Study Quality
Two reviewers independently completed a quality review for each study using a modified existing quality assessment tool.Reference Boyle 12 Quality scores were determined from eight key questions pertaining to sample representativeness, condition assessment, and statistical methods. Each study was given a quality score of 0 to 8 based on fulfillment of the quality criteria, as previously described.Reference Mah, Korngut, Dykeman, Day, Pringsheim and Jette 2
Statistical Analysis
Estimates of point and period prevalence can vary widely depending on the natural history of a disease; in the current study, both forms of prevalence were considered together because of the irreversible nature of muscular dystrophy. The pooled prevalence of combined dystrophies including Duchenne, Becker, congenital, facioscapulohumeral, limb girdle, myotonic overall and myotonic, facioscapulohumeral, limb girdle, Emery-Dreifuss, and congenital muscular dystrophies by age group per 100,000 were calculated where appropriate. Estimates using total population (males and females) as the denominator were considered separately from those including only children in the denominator.
To be included in the meta-analysis, studies needed to report the number of cases and sample size, the estimate with accompanying confidence intervals, or the information required to calculate the missing values. To assess for significant between-study heterogeneity, the Cochrane Q statistic was calculated and I 2 was used to quantify the magnitude of between-study heterogeneity. An a priori decision was made to use a random effects model because of the heterogeneous nature of the condition. Publication bias was investigated visually using funnel plots and statistically using Begg’s and Egger’s tests. For all tests, a p value less than 0.05 was deemed to be statistically significant. All statistical analyses were carried out in R (version 2.14) 13 ; the meta package (version 1.6-1.2010) was used to produce the stratified analyses and forest plots. 14 The metafor package was used to produce the pooled estimates using restricted maximum likelihood estimation.Reference Viechtbauer 15
Results
Identification and Description of Studies
The results of the combined search strategy yielded a total of 1104 citations; 167 articles met the criteria for full-text review. Thirty-one studies met all eligibility criteria and were included in the systematic review (Figure 1). Twenty-four studies were related to other muscular dystrophies apart from Duchenne and Becker muscular dystrophies.
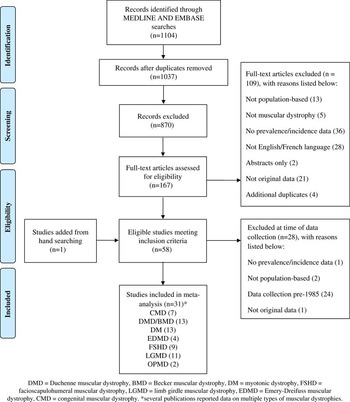
Figure 1 Flow chart of study selection.
Systematic Review of Muscular Dystrophies
Sixteen of the studies were conducted in Europe, including Darin and Tulinius,Reference Darin and Tulinius 16 De Munain et al,Reference López de Munain, Blanco, Emparanza, Poza, Martí Massó and Cobo 17 Fanin et al,Reference Fanin, Duggan, Mostacciuolo, Martinello, Freda and Sorarù 18 , Reference Fanin, Nascimbeni, Fulizio and Angelini 19 Hughes et al,Reference Hughes, Hicks, Nevin and Patterson 20 Magee and Nevin,Reference Magee and Nevin 21 Medica et al,Reference Medica, Marković and Peterlin 22 Mladenovic et al,Reference Mladenovic, Pekmezovic, Todorovic, Rakocevic-Stojanovic, Savic and Romac 23 Mostacciuolo et al,Reference Mostacciuolo, Miorin, Martinello, Angelini, Perini and Trevisan 24 , Reference Mostacciuolo, Pastorello, Vazza, Miorin, Angelini and Tomelleri 25 Norwood et al,Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 Siciliano et al,Reference Sposìto, Pasquali, Galluzzi, Rocchi, Solìto and Soragna 28 Sposito et al; Stensland et al,Reference Stensland, Lindal, Jonsrud, Torbergsen, Bindoff and Rasmussen 29 Urtasun et al,Reference Urtasun, Sáenz, Roudaut, Poza, Urtizberea and Cobo 30 and van der Kooi et al.31 Three studies were from the Middle East, including Blumen et al,Reference Blumen, Nisipeanu, Sadeh, Asherov, Blumen and Wirguin 32 El-Tallawy et al,Reference El-Tallawy, Khedr, Qayed, Helliwell and Kamel 33 and Segel et alReference Segel, Silverstein, Lerer, Kahana, Meir and Sagi 34 ; three studies were from Asia, including Chung et al,Reference Chung, Wong and Ip 35 Hsiao et al,Reference Hsiao, Chen, Li, Chiang, Lin and Pan 36 and Nakagawa et alReference Nakagawa, Nakahara, Yoshidome, Suehara, Higuchi and Fujiyama 37 ; one study by Ford et al was from New ZealandReference Ford, Kidd and Hammond-Tooke 38 ; and one study by Flanigan et al was from North America.Reference Flanigan, Coffeen, Sexton, Stauffer, Brunner and Leppert 39 All 24 studies reported on the prevalence of muscular dystrophy; no studies reported on its incidence. The studies were hospital- or clinic-based estimates of the prevalence of muscular dystrophies, with the exception of one door-to-door home-based survey by El-Tallawy et al in 2005.Reference El-Tallawy, Khedr, Qayed, Helliwell and Kamel 33
The diagnostic criteria for muscular dystrophies were based on predefined criteria including the age of onset and distribution of muscle weakness, associated symptoms, rate of disease progression, family history, serum creatine kinase, and histological studies, with or without neurophysiology tests such as electromyography and nerve conduction studies (Tables 1-6). Genetic data were not specified in earlier studies by Hughes et al,Reference Hughes, Hicks, Nevin and Patterson 20 Darin and Tulinius,Reference Darin and Tulinius 16 Chung et al,Reference Chung, Wong and Ip 35 El-Tallawy et al,Reference El-Tallawy, Khedr, Qayed, Helliwell and Kamel 33 Mostacciuolo et al,Reference Mostacciuolo, Miorin, Martinello, Angelini, Perini and Trevisan 24 Nakagawa et al,Reference Nakagawa, Nakahara, Yoshidome, Suehara, Higuchi and Fujiyama 37 van der Kooi et al,Reference van der Kooi, Barth, Busch, de Haan, Ginjaar and van Essen 31 and Blumen et al.Reference Blumen, Nisipeanu, Sadeh, Asherov, Blumen and Wirguin 32 Molecular genetic testing was used for diagnosis in some but not all patients in other studies by De Munain et al,Reference López de Munain, Blanco, Emparanza, Poza, Martí Massó and Cobo 17 Ford et al,Reference Ford, Kidd and Hammond-Tooke 38 Hsiao et al,Reference Hsiao, Chen, Li, Chiang, Lin and Pan 36 Magee & Nevin,Reference Magee and Nevin 21 Medica et al,Reference Medica, Marković and Peterlin 22 Segel et al,Reference Segel, Silverstein, Lerer, Kahana, Meir and Sagi 34 Siciliano et al,Reference Siciliano, Manca, Gennarelli, Angelini, Rocchi and Ludice 27 and Urtasun et al.Reference Urtasun, Sáenz, Roudaut, Poza, Urtizberea and Cobo 30 Genetic confirmation in most or all patients was reported by Norwood et al,Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 Mladenovic et al,Reference Mladenovic, Pekmezovic, Todorovic, Rakocevic-Stojanovic, Savic and Romac 23 Fanin et al,Reference Fanin, Duggan, Mostacciuolo, Martinello, Freda and Sorarù 18 Fanin et al,Reference Fanin, Nascimbeni, Fulizio and Angelini 19 Stensland et al,Reference Stensland, Lindal, Jonsrud, Torbergsen, Bindoff and Rasmussen 29 Sposito et al,Reference Sposìto, Pasquali, Galluzzi, Rocchi, Solìto and Soragna 28 and Mostacciuolo et al.Reference Mostacciuolo, Pastorello, Vazza, Miorin, Angelini and Tomelleri 25
Myotonic Dystrophy
Thirteen studies reported on the prevalence of myotonic dystrophy, and all were included in the meta-analysis. The majority of the studies did not distinguish between types 1 and 2 diseases. Eleven studies focused on all age groups, including De Munain et al,Reference López de Munain, Blanco, Emparanza, Poza, Martí Massó and Cobo 17 Ford et al,Reference Ford, Kidd and Hammond-Tooke 38 Hsiao et al,Reference Hsiao, Chen, Li, Chiang, Lin and Pan 36 Hughes et al,Reference Hughes, Hicks, Nevin and Patterson 20 Magee and Nevin,Reference Magee and Nevin 21 Medica et al,Reference Medica, Marković and Peterlin 22 Mladenovic et al,Reference Mladenovic, Pekmezovic, Todorovic, Rakocevic-Stojanovic, Savic and Romac 23 Nakagawa et al,Reference Nakagawa, Nakahara, Yoshidome, Suehara, Higuchi and Fujiyama 37 Norwood et al,Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 Segel et al,Reference Segel, Silverstein, Lerer, Kahana, Meir and Sagi 34 and Siciliano et al.Reference Siciliano, Manca, Gennarelli, Angelini, Rocchi and Ludice 27 Studies by Darin and TuliniusReference Darin and Tulinius 16 and Chung et alReference Chung, Wong and Ip 35 involved children only (Figure 2). The pooled prevalence of myotonic dystrophy in all age groups was 8.26 per 100,000 (95% confidence interval [CI], 4.99-13.68), and 1.41 per 100,000 (95% CI, 0.11-17.85) in children alone. Significant heterogeneity existed among studies of all age groups (I 2=99.1%, p<0.0001) and between the two pediatric studies (I 2=96.2%, p<0.0001).

Figure 2 Forest plots of individual studies and pooled prevalence estimates of myotonic dystrophy (DM).
Facioscapulohumeral Dystrophy
Nine studies reported on the prevalence of facioscapulohumeral dystrophy and all were included in the meta-analysis. Seven studies reported on all age groups, including El-Tallawy et al,Reference El-Tallawy, Khedr, Qayed, Helliwell and Kamel 33 Flanigan et al,Reference Flanigan, Coffeen, Sexton, Stauffer, Brunner and Leppert 39 Hughes et al,Reference Hughes, Hicks, Nevin and Patterson 20 Mostacciuolo et al,Reference Mostacciuolo, Pastorello, Vazza, Miorin, Angelini and Tomelleri 25 Nakagawa et al,Reference Nakagawa, Nakahara, Yoshidome, Suehara, Higuchi and Fujiyama 37 Norwood et al,Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 and Sposito et al.Reference Sposìto, Pasquali, Galluzzi, Rocchi, Solìto and Soragna 28 Two studies by Darin and TuliniusReference Darin and Tulinius 16 and Chung et alReference Chung, Wong and Ip 35 focused on children only (Figure 3). The pooled prevalence of facioscapulohumeral dystrophy in all age groups was 3.95 per 100,000 (95% CI, 2.89-5.40). The pooled prevalence in children only was 0.29 per 100,000 (95% CI, 0.03-3.00). Significant heterogeneity was present for both groups (I 2=89.5%, p<0.0001 for all ages, and I 2=77.1%, p=0.04 for children only).

Figure 3 Forest plots of individual studies and pooled prevalence estimates of facioscapulohumeral muscular dystrophy (FSHD).
Limb Girdle Muscular Dystrophy
Eleven studies reported on the prevalence of limb girdle muscular dystrophy and were included in the meta-analysis: nine reported on all age groups, including El-Tallawy et al,Reference El-Tallawy, Khedr, Qayed, Helliwell and Kamel 33 Fanin et al,Reference Fanin, Duggan, Mostacciuolo, Martinello, Freda and Sorarù 18 , Reference Fanin, Nascimbeni, Fulizio and Angelini 19 Hughes et al,Reference Hughes, Hicks, Nevin and Patterson 20 Nakagawa et al,Reference Nakagawa, Nakahara, Yoshidome, Suehara, Higuchi and Fujiyama 37 Norwood et al,Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 Stensland et al,Reference Stensland, Lindal, Jonsrud, Torbergsen, Bindoff and Rasmussen 29 Urtasun et al,Reference Urtasun, Sáenz, Roudaut, Poza, Urtizberea and Cobo 30 and van der Kooi et al.Reference van der Kooi, Barth, Busch, de Haan, Ginjaar and van Essen 31 Two studies by Darin and TuliniusReference Darin and Tulinius 16 and Chung et alReference Chung, Wong and Ip 35 focused on children alone (Figure 4). Among studies including all age groups, the pooled prevalence of limb girdle muscular dystrophy was 1.63 per 100,000 (95% CI, 0.94-2.81). In children alone, the pooled prevalence was 0.48 per 100,000 (95% CI, 0.18-1.31). Significant heterogeneity was present for studies reporting on all age groups (I 2=96.3%, p<0.0001), but not for those including children only (I 2=44.4%, p=0.18).

Figure 4 Forest plots of individual studies and pooled prevalence estimates of limb girdle muscular dystrophy (LGMD).
Emery-Dreifuss Muscular Dystrophy
Four studies reported on the prevalence of Emery-Dreifuss muscular dystrophy. Three studies reported on all age groups and were included in the meta-analysis, including El-Tallawy et al,Reference El-Tallawy, Khedr, Qayed, Helliwell and Kamel 33 Hughes et al,Reference Hughes, Hicks, Nevin and Patterson 20 and Norwood et al.Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 Chung et alReference Chung, Wong and Ip 35 was the only pediatric study, with a prevalence of 0.22 per 100,000 children. The pooled prevalence of Emery-Dreifuss muscular dystrophy in all age groups was 0.39 per 100,000 (95% CI, 0.12-1.30; Figure 5), with significant heterogeneity among the estimates (I 2=71.3%, p=0.03).

Figure 5 Forest plots of individual studies and pooled prevalence estimates of Emery-Dreifuss muscular dystrophy (EDMD).
Congenital Muscular Dystrophy
Four studies reported on the prevalence of congenital muscular dystrophy in all age groups, including El-Tallawy et al,Reference El-Tallawy, Khedr, Qayed, Helliwell and Kamel 33 Hughes et al,Reference Hughes, Hicks, Nevin and Patterson 20 Nakagawa et al,Reference Nakagawa, Nakahara, Yoshidome, Suehara, Higuchi and Fujiyama 37 and Norwood et al.Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 Three studies reported on the prevalence in children alone, including Darin and Tulinius,Reference Darin and Tulinius 16 Chung et al,Reference Chung, Wong and Ip 35 and Mostacciuolo et alReference Mostacciuolo, Miorin, Martinello, Angelini, Perini and Trevisan 24 (Figure 6). The pooled prevalence of congenital muscular dystrophy in all age groups was 0.99 per 100,000 (95% CI, 0.62-1.57), and 0.82 per 100,000 (95% CI, 0.27-2.47) in children only. Significant heterogeneity was present among the pediatric studies (I 2=87.4%, p=0.0003) but not for studies reporting on all age groups (I 2=55.5%, p=0.08).

Figure 6 Forest plots of individual studies and pooled prevalence estimates of congenital muscular dystrophy (CMD).
Oculopharyngeal Muscular Dystrophy
Two studies described the prevalence of oculopharyngeal muscular dystrophy in all age groups, including Blumen et alReference Blumen, Nisipeanu, Sadeh, Asherov, Blumen and Wirguin 32 and Norwood et al.Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 A pooled analysis was not performed because of the limited number of studies.
Combined Muscular Dystrophies
Five studies reported on the overall prevalence of combined muscular dystrophies, including Duchenne muscular dystrophy, Becker muscular dystrophy, congenital muscular dystrophy, facioscapulohumeral dystrophy, limb girdle muscular dystrophy, and myotonic dystrophy by Chung et al,Reference Chung, Wong and Ip 35 Darin and Tulinius,Reference Darin and Tulinius 16 Hughes et al,Reference Hughes, Hicks, Nevin and Patterson 20 Nakagawa et al,Reference Nakagawa, Nakahara, Yoshidome, Suehara, Higuchi and Fujiyama 37 and Norwood et al.Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 The overall pooled prevalence of combined muscular dystrophies was 16.14 per 100,000 (95% CI, 11.21-23.23). Random effects model showed significant heterogeneity, with I 2=97.5%, p<0.0001 (Figure 7).

Figure 7 Forest plots of individual studies and pooled prevalence estimates of the combined muscular dystrophies (MDs).
Publication Bias
Using both Begg’s and Egger’s tests, no evidence of publication bias was found for the muscular dystrophies (p>0.05), apart from facioscapulohumeral dystrophy. There was evidence of publication bias based on Egger’s test for facioscapulohumeral dystrophy (p=0.008), but not Begg’s (p=0.13); visual inspection of the funnel plot revealed an asymmetry, suggesting the potential of missing studies with a lower prevalence.
Study Quality
As indicated in Table 7, there were notable differences in research methodology as well as study quality. The median study quality score for studies reporting on the prevalence of muscular dystrophies was 7 of 8 (range, 4-8). All 31 articles described the target population in detail and 27 of 31 studies sampled either the entire population or used probability sampling. Of those studies necessitating a response rate, 26 reported a response rate greater than 70% and described the nonresponders adequately. The majority of studies reported on whether their sample was representative of the target population. All studies used standardized data collection methods and most used validated criteria to assess for the presence of muscular dystrophies. Only five studies reported estimates with their accompanying confidence intervals or by subgroups.
Discussion
According to Emery’s report in 1991, the prevalence of inherited neuromuscular disorders including Duchenne and Becker muscular dystrophies, spinal muscular atrophy, myotonic dystrophy, congenital myotonias, hereditary motor and sensory neuropathies, familial motor neuron disease, and familial myasthenia gravis was estimated to be around 28.6 per 100,000, or 1 in 3500.Reference Emery 40 In the current systematic review, the overall worldwide prevalence of combined muscular dystrophies is estimated to be 16.14 per 100,000, or 1 in 6200. The reports of muscular dystrophies prevalence included in this systematic review were all population-based, used appropriate methodology, and were of good quality. However, several outlier estimates were observed. The overall prevalence estimates for facioscapulohumeral dystrophy in Egypt was high, likely as a result of the relatively small population sampled, the door-to-door survey methodology, and the number of cases identified.Reference El-Tallawy, Khedr, Qayed, Helliwell and Kamel 33 Also, the overall prevalence estimate for myotonic dystrophy type 1 in Spain of 26.53 per 100,000 is higher than in the other included studies without any clear methodological differences, possibly related to a founder effect.Reference López de Munain, Blanco, Emparanza, Poza, Martí Massó and Cobo 17 In contrast, a lower prevalence estimate of myotonic dystrophy was observed in China and Taiwan (0.37 and 0.46 per 100,000, respectively); this may be attributed to variation in the availability of genetic testing for confirmation of diagnosis and restriction of the sample population to those younger than 19 years of age.Reference Chung, Wong and Ip 35 , Reference Hsiao, Chen, Li, Chiang, Lin and Pan 36 Similarly, lower prevalence estimates were found in other systematic reviews of neurological conditions such as Huntington or Parkinson diseases in Asia.Reference Pringsheim, Wiltshire, Day, Dykeman, Steeves and Jette 41 , Reference Pringsheim, Jette, Frolkis and Steeves 42 However, Darin and Tulinius also restricted their sample population to those younger than 16 years of age and demonstrated a more typical prevalence estimate of myotonic dystrophy at 5.0 per 100,000.Reference Darin and Tulinius 16 Therefore, the variation may also be related to genetic differences and/or the influence of migration on the prevalence of the inherited muscular dystrophies.
Comparison of pooled estimates demonstrates that myotonic dystrophy type 1 is the most prevalent muscular dystrophy in childhood and in the overall population, with a pooled prevalence of 8.26 (95% CI, 7.99-13.68) per 100,000. Emery estimated an overall prevalence of 5.0 per 100,000 in adults.Reference Emery 40 Much higher prevalence of myotonic dystrophy type 1 had been reported because of founder effect and geographical isolation. A prevalence of 158 per 100,000 was recently reported by Mathieu et al in 2012 at the Saguenay-Lac-Saint-Jean region in Northern Quebec, Canada; the prevalence has progressively declined over the past two decades because of reduced fertility rate.Reference Mathieu and Prévost 43 Facioscapulohumeral dystrophy is the second most prevalent muscular dystrophy; the pooled prevalence in all age groups from this study was 3.95 (95% CI, 2.89-5.40) per 100,000, which is about twice as high as the estimate of 2 per 100,000 by Emery in 2001.Reference Emery 40 Duchenne muscular dystrophy remains the most prevalent muscular dystrophy of childhood in boys. The estimated prevalence of Duchenne and Becker muscular dystrophies according to Emery was 6.3 and 2.4 per 100,000, respectively, which is similar to our recent estimates of 4.78 (95% CI, 1.94-11.81) and 1.53 (95% CI: 0.26-8.94) per 100,000.Reference Mah, Korngut, Dykeman, Day, Pringsheim and Jette 2
Variation in the reported prevalence of muscular dystrophies could be related to genetic variations between populations or ethnic groups as well as differences in available molecular diagnostic tools for accurate diagnosis.Reference Chung, Wong and Ip 35 , Reference Hsiao, Chen, Li, Chiang, Lin and Pan 36 Furthermore, neuromuscular disorders are relatively uncommon in the general population, and one must often resort to hospital charts and other sources of medical information in the community for the number of diagnosed cases. The availability of such information varies among countries and within regions. As well, some affected individuals may not seek medical attention; undiagnosed cases therefore may lead to an underestimation of the true prevalence of muscular dystrophies. On the other hand, advances in research activity may generate an increased awareness in a region for a particular group of disorders, as may be seen by Norwood et al within the United Kingdom.Reference Norwood, Harling, Chinnery, Eagle, Bushby and Straub 26 Comparatively fewer cases of muscular dystrophy were diagnosed during the first 4 years of life, as indicated in a recent study on congenital myotonic dystrophyReference Campbell, Levin, Siu, Venance and Jacob 44 ; diagnostic difficulties may contribute to the lower prevalence of muscular dystrophies as reported in early childhood. Prevalence figures in school-aged children are therefore likely to give a better estimate of the true childhood prevalence of neuromuscular disorders.Reference Darin and Tulinius 16
As previously mentioned,Reference Mah, Korngut, Dykeman, Day, Pringsheim and Jette 2 limitations of this systematic review include variability in methodology, nonrandom geographic distribution, and the lack of validated international classification of disease codes for the muscular dystrophies among the studies.Reference St. Germaine-Smith, Metcalfe, Pringsheim, Roberts, Beck and Hemmelgarn 45 The lack of genetic testing in earlier studies may also lead to an imprecise estimation of the true prevalence of the muscular dystrophies. It was not possible to calculate a pooled estimate of myotonic dystrophy type 2 as several of the epidemiologic studies were conducted prior to the discovery of the gene for type 2 disease.Reference Ranum, Rasmussen, Benzow, Koob and Day 4 There was evidence of publication bias, but only for facioscapulohumeral dystrophy. Flanigan et al reported a high prevalence of disease in a geographically and genetically restricted populationReference Flanigan, Coffeen, Sexton, Stauffer, Brunner and Leppert 39 ; there may be other studies with a lower prevalence of facioscapulohumeral dystrophy that were not identified in our search. Despite great efforts to ensure the search strategy to be as comprehensive as possible, we did not include abstracts, gray literature, or articles written in languages apart from English or French. Application of these results must be made in light of these limitations. Multiple sources of case ascertainment beyond clinical-based settings such as national registries, genetic databases, and patient organizations should be considered for future incidence or prevalence studies of the muscular dystrophies.
Conclusion
This is a meta-analysis of the minimum prevalence estimates for myotonic dystrophy, facioscapulohumeral dystrophy, limb girdle muscular dystrophies, congenital muscular dystrophies, and combined muscular dystrophies derived from studies around the world. Population-based registries for the muscular dystrophies were limited to parts of Europe, Asia, and North America only. Substantial gaps exist with no available data from many other continents, limiting the generalization of the reports. In the United States, the reported national costs of illness for Duchenne muscular dystrophy, myotonic dystrophy, and other progressive neuromuscular diseases has been estimated to exceed more than $1 billion per year.Reference Larkindale, Yang, Hogan, Simon, Zhang and Jain 46 Our pooled estimates are useful for calculating projections of expected case numbers in regions without accurate prevalence data facilitating estimation of health care burden, economic impact, and clinical resource requirements. Clinical research must continue to be anchored to epidemiological understanding to enable interpretation of relevance to, and effect of, the overall patient population.
Table 1 Prevalence studies on myotonic dystrophy listed by study population and year of publication

±=with or without; CI=confidence interval; DM1=myotonic dystrophy type 1; DM2=myotonic dystrophy type 2; EMG=electromyography.
Table 2 Prevalence studies on facioscapulohumeral dystrophy listed by study population and year of publication

±=with or without; CI=confidence interval; EMG=electromyography.
Table 3 Prevalence studies on limb girdle muscular dystrophy listed by study population and year of publication

±=with or without; CI=confidence interval; EMG=electromyography.
Table 4 Prevalence studies on Emery-Dreifuss muscular dystrophy listed by study population and year of publication

±=with or without; CI=confidence interval; EMG=electromyography.
Table 5 Prevalence studies on congenital muscular dystrophy listed by study population and year of publication

±=with or without; CI=confidence interval; EMG=electromyography.
Table 6 Prevalence studies on oculopharyngeal muscular dystrophy listed by study population and year of publication

CI=confidence interval.
Table 7 Quality scores of studies included in the muscular dystrophies systemic review

Acknowledgments
This study is part of the National Population Health Study of Neurological Conditions. We wish to acknowledge the membership of Neurological Health Charities Canada and the Public Health Agency of Canada for their contribution to the success of this initiative. Funding for the study was provided by the Public Health Agency of Canada. The opinions expressed in this publication are those of the authors/researchers, and do not necessarily reflect the official views of the Public Health Agency of Canada. We would like to acknowledge Diane Lorenzetti for her assistance in developing the search strategies. Nathalie Jetté holds a Population Health salary award from Alberta Innovates Health Solutions and a Canada Research Chair Tier 2 in Neurological Health Services Research.
Disclosures
TP has served as a consultant for and received consulting fees from Teva Canada; received an unrestricted grant from Shire Canda; and been a meeting attendee and received support to attend meetings for Allergan Canada. NJ has received a research grant and research support paid to the university from the Public Health Agency of Canada; been an award recipient and received a salary award paid to the university from the Canada Research Chair Program; and received a salary award paid to the university from Albert Innovates Health Solutions. JM, KF, JD, LD, and LK have nothing to disclose.














