


Cerebral vasospasm is commonly reported following traumatic brain injury (TBI) and traumatic subarachnoid hemorrhage (tSAH). However, severe vasospasm after TBI with minimal, focal tSAH is rare and often forgotten.
A 62-year-old woman experienced an evident mechanical fall with head injury, but with no loss of consciousness or abrasions to the head or neck. She was being treated for hypertension and a previous unprovoked pulmonary embolism with trandolapril, hydrochlorothiazide, and dabigatran.
She initially presented with confusion, vomiting, and urinary incontinence. Glasgow Coma Scale was 14. She had no fever, meningismus, or focal neurological deficits on presentation. There were no prodromal symptoms, thunderclap headaches, or changes in cognition in the weeks or days leading to the head injury. Initial computed tomography (CT) and magnetic resonance imaging showed minimal convexal tSAH and minimal intraventricular hemorrhage (Figure 1). Magnetic resonance angiography (MRA) was normal. She was hyponatremic (Na, 126 mmol/L) and had leukocytosis (white blood cell count, 21.6 × 109/L), attributed to urinary tract infection, which quickly normalized to 10.6 × 109/L 4 days following her admission. Blood cultures were negative. She was treated for delirium secondary to hypovolemia and a presumed urinary tract infection. A follow-up CT scan on day 7 showed expected evolution/stabilization of her bleed. Her delirium resolved by day 8, and the patient was prepared for discharge.
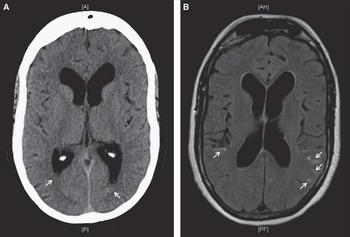
Figure 1 Initial computed tomography scan of the head (A) shows trace intraventricular hemorrhage in bilateral occipital horns of the lateral ventricles. Axial fluid-attenuated inversion recovery magnetic resonance imaging (B) revealed the presence of minimal convexal subarachnoid hemorrhage in the posterior aspect of both cerebral hemispheres (white arrows).
However, on day 10, the patient developed left hemiparesis, left homonymous hemianopsia, and neglect. CT/CT angiography (CTA) revealed a right parietal infarct and diffuse, bihemispheric vasospasm with no topographical relationship to the focal tSAH (Figure 2). No proximal or distal aneurysms were seen. A thorough infectious and inflammatory workup was negative at that time.
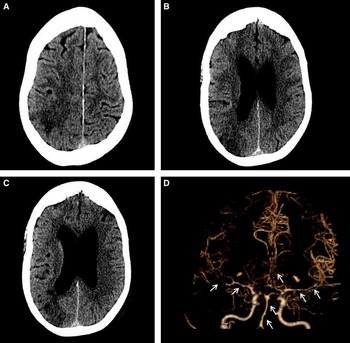
Figure 2 Imaging after sudden onset of new focal neurological symptoms. Computed tomography scan of the head (A-C) shows evolving subacute ischemia in the right posterior parietal and superior occipital lobe. Computed tomography angiogram (D) demonstrates diffuse cerebral vasospasm, most pronounced in the bilateral M1 segments and right M2 branches, but also present in the left M2, bilateral A1s and A2s, and the basilar artery. Vasospasm was also noted in the left V4 segment and bilateral P1s and P2s (not shown).
The patient was treated with milrinone 20 mg in 80 ml normal saline infused at 0.75 mg/kg/min, nimodipine 60 mg by mouth every 4 hours, and magnesium sulfate 5 g/250 ml saline over 5 hours. Response to treatment was monitored with serial transcranial Doppler ultrasounds and CTAs. The transcranial Doppler ultrasounds were interpreted by a single radiologist using sonographic criteria. The CTAs were read by two separate radiologists, who used coronal, sagittal, maximum intensity projection, and three-dimensional reformatted images and volumetric measurements. Vasospasm resolved by day 21, at which time the patient was completely compos mentis but had persistent neurological deficits relevant to her infarct. She was transferred to inpatient rehabilitation and achieved independence for basic activities of daily living.
Cerebral vasospasm is a reported complication of TBI and tSAH. Its incidence appears to correlate with the severity of bleeding,Reference Oertel, Boscardin and Obrist 1 and it is rarely reported following a relatively small subarachnoid hemorrhage.
In our patient, TBI and tSAH were relatively minor and focal. It was therefore unexpected for her to suffer such severe bilaterally diffuse cerebral vasospasm. An intracranial or systemic infection or inflammatory process with associated vasculopathy was ruled out given the lack of fever, other constitutional symptoms, meningismus, or prodromal state. She also had a negative thorough inflammatory screen and more importantly had regained compos mentis without any targeted therapies for such etiologies. She also never experienced any new or thunderclap headaches, was not exposed to any vasoactive substances, and the precipitant fall was clearly mechanical, rendering reversible cerebral vasoconstriction syndrome unlikely at initial presentation to hospital.
The pathophysiology of cerebral vasospasm after TBI and subarachnoid hemorrhage remains elusive. Accepted theories suggest that blood initiates signaling pathways that upregulate the production of vasoconstrictors and inhibit vasodilators. Greater blood product deposition increases the risk of vasospasm compared with injuries with less subarachnoid hemorrhage.Reference O’Brien, Maa and Yeates 2 , Reference Pluta 3 However, tSAH is not required for vasospasm in TBI. We hypothesize that the vasospasm was largely due to TBI independent of the bleeding. Fittingly, a study of blast-induced TBI suggested that acute, blast-like injury alone could potentiate cerebral vasospasm. In this case, blast-wave associated pressure pulses may initiate cerebral vasospasm within 24 hours of injury by inducing short-term endothelin-1 hypersensitivity, prolonged hypercontractility, and maladaptive vascular remodeling.Reference Alford, Dabiri, Goss, Hemphill, Brigham and Parker 4 Other suggested mechanisms for TBI-induced vasospasm in the absence of bleeding include direct mechanical irritation or stretching, which may lead to mechanical alteration of cerebral blood vessels. Furthermore, the disruption of neuronal cell membranes in traumatized brain parenchyma can lead to the release of potent vasoactive substances which may induce or exacerbate vasospasm.Reference Perrein, Petry, Reis, Baumann, Mertes and Audibert 5
Our patient presented at 10 days after her initial TBI; TBI- or tSAH-associated vasospasm typically occur within 48 hours of the initial event.Reference Perrein, Petry, Reis, Baumann, Mertes and Audibert 5 It is possible that vasospasm was present earlier, but not detected until subsequent neurologic deficits occurred as a result of infarction. Although MRA is sensitive for detecting proximal areas of stenosis/vasospasm, it can miss distal pathology. Accordingly, it is conceivable that distal regions of vasospasm may have been missed on the initial MRA and otherwise been detected had a CTA been performed instead at that time.
Evidence-based treatment options for vasospasm following TBI and tSAH are lacking. Some have suggested endovascular therapy with balloon angioplasty or infusion of vasodilators. Nimodipine may be beneficial, and other calcium channel blockers such as verapamil and nicardipine have been proposed. The Montreal Neurological Hospital protocol was also associated with good functional outcomes in a large case series of 88 patients. In this case, milrinone is administered while maintaining euvolemia, eunatremia, euglycemia, and normotension. Of particular note, however, is that current evidence does not support the use of calcium channel blockers as a prophylactic agent post-TBI.Reference Perrein, Petry, Reis, Baumann, Mertes and Audibert 5 , Reference Lannes, Teitelbaum, del Pilar Cortés, Cardoso and Angle 6
Our patient demonstrates that TBI with even minimal tSAH can be followed by widespread cerebral vasospasm and infarction. Physicians should consider cerebral vasospasm as a potential mechanism in patients who suffer a stroke following any level of TBI. Whether or not close monitoring with serial imaging can help detect early vasospasm and, more importantly, improve outcomes of patients with any degree of TBI by preventing cerebral infarction requires further exploration.
Disclosures
The authors have no disclosures to report.


