


Heterogeneity is a key feature of the hereditary spastic paraplegias (HSPs). Indeed, to date, autosomal, sex-linked, and cytoplasmic inheritance have been reported, an ample array of complicated phenotypes disclosed, roughly 60 loci mapped, and over 40 disease genes clonedReference Finsterer, Löscher, Quasthoff, Wanschitz, Auer-Grumbach and Stevanin 1 (see also www.genenames.org/ for the full list of the SPG genes). In common practice, the gene-after-gene testing strategy allows definition of the molecular basis in about half of HSP cases,Reference Finsterer, Löscher, Quasthoff, Wanschitz, Auer-Grumbach and Stevanin 1 unless peculiar features emerge during examination or follow-up. Although assessment of the large panel of SPG genes is a novel opportunity afforded by new methods of next-generation sequencing (NGS),Reference Kumar, Blair and Vandebona 2 interpretation of the results remains challenging without the information that can be provided by full and careful neurological examination. Moreover, several clinical conditions said to mimic HSP, including spastic-ataxias and axonal peripheral neuropathies,Reference Finsterer, Löscher, Quasthoff, Wanschitz, Auer-Grumbach and Stevanin 1 might be allelic to new SPG genes, making it even more complicated to provide the final molecular diagnosis, especially in isolated cases.
We describe a sporadic HSP patient whose severe and exclusive pyramidal tract involvement initially prompted inappropriate genetic testing. Careful consideration of her clinical presentation eventually led to diagnosis of a frataxin-related disorder.
Patients and Methods
A 34-year-old Italian woman, with an unremarkable family history, reported leg stiffness and clumsiness after her first pregnancy at the age of 27 and worsening of these symptoms during a second pregnancy at the age of 30. Subsequently, her clinical condition deteriorated further, leading to loss of independent walking at the age of 32. The patient had unremarkable prenatal, perinatal, and early childhood histories, although three episodes of peripheral VII nerve palsy, occurring at the ages of 6, 11 and 30 years and fully resolving with steroid therapy, were reported. The patient’s medical history was also significant for remittent Graves’ disease, for which she was being treated with methimazole (15 mg/day). Because of her progressive clinical complaints, she was referred to our attention (at the Stella Maris neurological center) for a second opinion, suspected of being affected by Strumpell-Lorrain disease.
Neurological examination (see supplemental video) at the age of 34 years showed a clearly spastic gait with leg adduction and slight knee flexion. Ambulation was slow and possible only with unilateral support (a cane). The patient was not able to jump or run, or walk on her heels or toes. Her strength was significantly reduced (Medical Research Council grading 3+) and her muscle tone markedly increased in the lower but not in the upper limbs, whereas deep tendon reflexes were enhanced in the four limbs, with ankle clonus and bilateral Babinski signs. Proprioception and vibration sensation (ankle and knee) were also mildly reduced in the lower limbs. The cranial nerves were spared and she displayed neither nystagmus nor oculomotor abnormalities except for minimal fragmentation of smooth pursuit. Finger-to-nose and heel-to-knee tests were normal. Subjective imbalance was reported by the patient especially when changing direction and standing with eyes closed. The Romberg maneuver was, however, negative. The patient also experienced occasional urinary urgency. She showed no cognitive, language, or dysarthria, or skeletal abnormalities. Routine biochemistry, vitamin B12, vitamin E, venereal disease research laboratory test, chest radiograph, and electrocardiogram were also normal, although persistently high levels of serum iron were found. Her Spastic Paraplegia Rating Scale Annex 1 (SPRS) score was 23/52 and her Scale for Assessment and Rating of Ataxia score was 3/40 (only gait and stance). Brain magnetic resonance imaging (MRI) (Figure a), at age 34 years, displayed slight enlargement of the pericerebellar spaces in both the vermis and the hemispheres, whereas a spine MRI (Figure b) showed questionable cervical cord atrophy and some thinning of the dorsal tract. Motor evoked potentials showed delayed central conduction time. Peripheral nerve conduction studies, and visual and brainstem evoked potentials were all unremarkable. Heart (echocardiogram) and retinal investigations (optical coherence tomography examination) were also normal.
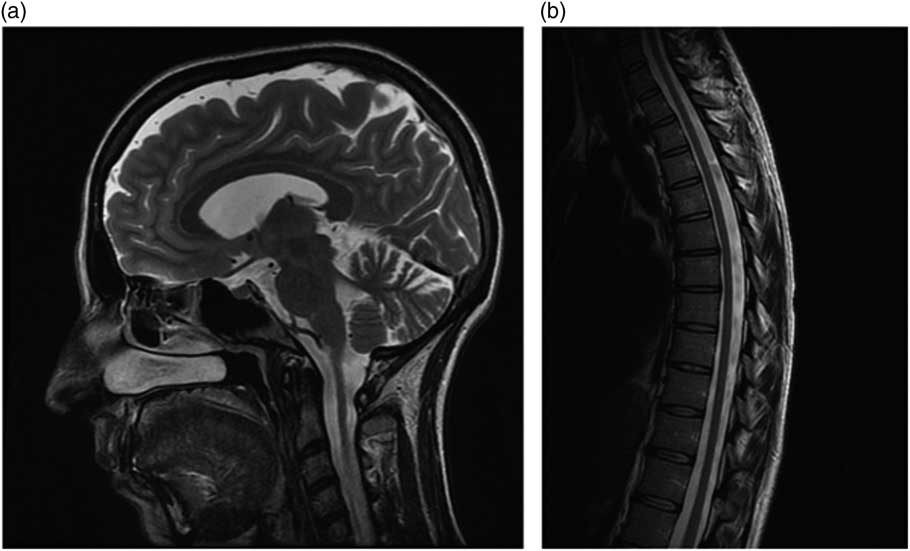
Figure Brain (a) and spine (b) MRI features in the patient.
Total genomic DNA was purified from peripheral blood and mutations in ATL1/SPG3A, SPAST/SPG4, CYP7B1/SPG5, PGN/SPG7, KIF5A/SPG10, RTN2/SPG12 and REEP1/SPG31 were all ruled out using polymerase chain reaction-based sequencing and multiplex ligation-dependent probe amplification methodologies. Conversely, based on a clinical guess, we detected the pathological GAA expansion in the first intron of FXN, the frataxin gene (GAA1 and GAA2 expansions consisted of 164 and 1041 uninterrupted repeats, respectively). This finding is consistent with a molecular diagnosis of Friedreich’s ataxia with retained tendon reflexes.Reference Delatycki and Corben 3
Discussion
Friedreich’s ataxia (FRDA) is the most common autosomal recessive hereditary ataxia with a prevalence of 1:50 000 in the European population.Reference Delatycki and Corben 3 , Reference Parkinson, Boesch, Nachbauer, Mariotti and Giunti 4 Harding’s criteria for typical FRDA are: onset before 25 years, progressive ataxia, absent knee and ankle jerks, extensor plantar response, sensory axonal neuropathy and, after five years, dysarthria. The pathological GAA expansion in FXN results in decreased expression of frataxin, a mitochondrial protein involved in the biogenesis of iron-sulfur clusters and the accumulation of iron in mitochondria. The length of GAAs is directly correlated with disease severity and progression, age at onset, and the presence of additional co-morbidities, including cardiomyopathy, diabetes mellitus, scoliosis, hearing loss and visual loss.Reference Parkinson, Boesch, Nachbauer, Mariotti and Giunti 4
Atypical presentations, such as the one we observed in our patient, who showed a progressive spastic paraparesis without gait or limb ataxia, are rare in large FRDA series,Reference Parkinson, Boesch, Nachbauer, Mariotti and Giunti 4 usually related to late-onset and smaller GAA1 size, and difficult to diagnose without a positive family history. A few similar cases have been described previously in the literature, prior to the identification of the first SPG genes,Reference Finsterer, Löscher, Quasthoff, Wanschitz, Auer-Grumbach and Stevanin 1 including a spastic variant of the Acadian form of FRDA described in the early 1980s.Reference Barbeau, Roy, Sadibelouiz and Wilensky 5 With hindsight, the spinal cord thinning, together with the slight enlargement of the cerebellar sulci observed in our patient, might have been taken as “red flags” pointing to a variant of FRDA and making it possible to avoid time-consuming analyses of several HSP genes.
Of note, having received the correct diagnosis, the patient was put on an idebenone (150 mg/day), ubidecarenone (1.5 g/day) and baclofen (50 mg/day) combined therapy. She also receives follow-up for her co-morbid conditions. Her latest SPRS score, at age 35, has slightly improved (19/52) especially in items related to weakness and pain.
Conclusion
The present case highlights the value of the “old” recommendation of an accurate differential diagnosis in isolated cases of HSP. This should consider clinical conditions that can present in a similar manner, including B12 deficiency, L-dopa responsive dystonia, structural spinal cord disorders, spinocerebellar ataxias, and lysosomal enzyme deficits. Disorders, the prognosis of which differ significantly from HSP, such as multiple sclerosis and motor neuron disease, should also be excluded. This especially applies when family history is not sufficiently informative,Reference Finsterer, Löscher, Quasthoff, Wanschitz, Auer-Grumbach and Stevanin 1 even though it is tempting to turn to NGS strategies for a rapid molecular definition. All the clinical tests, including the relatively cheap assay for pathological GAA expansion, if wisely used, have the capacity to direct more appropriately genetic screening which, whether using old or modern methods, is usually more demanding. The results of these tests can eventually modify clinical care and follow-up approaches, as in the case we described, as well as current and future therapeutic opportunities.
This clinical case deserves a final comment: The high frequency of cranial nerve palsy observed in our patient, long before the onset of her movement disorder, is intriguing. Although auditory and visual neuropathy have been described in FRDAReference Delatycki and Corben 3 , VII cranial nerve palsy seems, however, to be unrelated to the neurodegenerative disorder manifesting in our patient. In addition, it is unclear why the clinical manifestations in this patient mainly affected the pyramidal tract. It is tempting to hypothesize in this patient an effect of pathological GAA expansion on oxidative stressReference Delatycki and Corben 3 resembling those associated with mutations in paraplegin and AFG3L2, two known causes of spastic-ataxia, acting as regulators of the proof-reading activity on inner mitochondrial membrane proteins.Reference Finsterer, Löscher, Quasthoff, Wanschitz, Auer-Grumbach and Stevanin 1
Acknowledgements and Funding
The authors thank Catherine J. Wrenn for her editorial assistance.
This study was funded in part by Telethon Italy (grant GPP10121A to FMS).
Disclosures
The authors have no conflicts of interest to disclose.
Supplementary material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/cjn.2014.28


