


The overall aim of the present study is to attempt to identify robust and predictive markers, or patterns or clusters of markers, which can be used to assess inflammation in human nutrition studies in the general population. Inflammation is a normal component of host defence, initiating the mechanisms involved in pathogen killing and protection against other insults. Thus, in a physiological context, inflammation is protective. Typically, the inflammatory response is activated rapidly in response to infection or another trigger and then follows a temporal pattern of cellular activation and chemical mediator release. Once the infection or the other insult is eliminated, or at least controlled, mechanisms come into play to terminate inflammation in order to limit further damage to the host and to initiate tissue repair. This process is termed resolution of inflammation, and it is now recognised to be an active process involving specific mediators that act to down-regulate the processes that were earlier activated. Failure to resolve inflammation may permit the normally acute inflammatory processes to become chronic. Chronic inflammation may also be driven by continuous exposure to the triggering agent. Chronic inflammation is a well-recognised component of many pathologies and is a target for many pharmacologic interventions. In order to examine the role of diet, foods or specific nutrients on inflammation, it is necessary to identify candidate biomarkers. This review describes a systematic approach to identify generic biomarkers of inflammation and to consider the factors that may influence the status of those biomarkers.
Biomarkers of inflammation
Inflammation: a general overview
Inflammation is a normal host defence mechanism that protects the host from infection and other insults; it initiates pathogen killing, as well as tissue repair processes, and helps to restore homeostasis at infected or damaged sites(Reference Calder, Albers and Antoine1). A number of different triggers can initiate inflammation. These triggers include the presence of microbial products, tissue damage and metabolic stress (Fig. 1). Exposure to the trigger may involve a breakdown in barrier function or a loss of normal immune tolerance. Irrespective of the nature of the trigger, a common set of cellular pathways is initiated(Reference Sun, Ji and Kersten2). These pathways include activation of signalling via Toll-like receptors and nuclear factor (NF)-κB, formation of a multi-protein complex termed the inflammasome that is responsible for the release of several inflammatory cytokines, and endoplasmic reticulum (ER) stress (Fig. 1)(Reference Sun, Ji and Kersten2). The resulting inflammatory response is typified by redness, swelling, heat, pain and loss of function, and involves interactions among many cell types and the production of, and responses to, a vast number of chemical mediators. The precise timing and vigour of the inflammatory response, including the exact cells and mediators involved, is determined by the nature, extent and location of the trigger. These factors act to shape and maintain the inflammatory response. Once the infection or the other trigger is eliminated, or at least controlled, mechanisms come into play to terminate inflammation in order to limit further damage to the host and to initiate tissue repair. This self-regulating process is termed resolution of inflammation and involves the activation of negative feedback mechanisms such as the secretion of anti-inflammatory cytokines, the inhibition of pro-inflammatory signalling cascades, the shedding of receptors for inflammatory mediators and the activation of regulatory cells. Properly controlled inflammation is essential to remain healthy and maintain homeostasis, but inflammation that involves a loss of tolerance or regulatory processes may become pathological(Reference Calder, Albers and Antoine1).

Fig. 1 Schematic overview of the common inflammatory response initiated by different triggers. Triggers are those factors that can directly initiate an inflammatory response. ER, endoplasmic reticulum; TLR, Toll-like receptor; TNFR, TNF receptor.
Inflammation may be classified as acute or chronic (Table 1). Acute inflammation is the initial response of the body to an infectious agent or another inflammatory trigger (e.g. tissue damage by wounding or irradiation) and is achieved by the increased movement of plasma and leucocytes (especially granulocytes) from the blood into the site of infection or injury. A cascade of biochemical events matures and propagates the inflammatory response, involving the local vascular system, the immune system and various cells within the injured tissue. Acute inflammation is typically self-limiting with resolution being activated once the cause of the initial response is contained or eliminated. Resolution is itself an active process involving specific cell types and specific anti-inflammatory cytokines(Reference Iyer and Cheng3) and pro-resolving lipid mediators(Reference Fredman and Serhan4, Reference Serhan and Savill5). Prolonged, or chronic, inflammation involves a progressive shift in the type of cells present at the site of inflammation and simultaneous destruction and healing of the tissue due to the ongoing inflammatory process. Inflammation may become pathological due to the loss of tolerance and/or of regulatory processes such as resolution. Where this becomes excessive, irreparable damage to host tissues and disease can occur(Reference Calder, Albers and Antoine1). Such diseases are characterised by markedly elevated concentrations of inflammatory markers and of activated inflammatory cells at the site of tissue damage and in the systemic circulation; this state of inflammation may be regarded as ‘high grade’. These diseases include rheumatoid arthritis (RA), inflammatory bowel diseases (IBD), atopic dermatitis, psoriasis and asthma. Chronic inflammation can also be of a ‘low grade’ and under such circumstances, overt clinical manifestations can be minimal or absent; the elevation in the concentrations of inflammatory markers and of inflammatory cells in the systemic circulation is not as great as that seen in the frank chronic inflammatory conditions described above. Low-grade asymptomatic inflammation can occur in adipose tissue as a feature of obesity(Reference Calder, Ahluwalia and Brouns6). Under these conditions, the adipocyte itself becomes the source of inflammation-associated adipokines, although there is also infiltration of adipose tissue by macrophages and T cells, which make important contributions to the inflammatory output from adipose tissue(Reference Conde, Scotece and Gomez7). The cytokines and adipokines released probably contribute to the insulin resistance seen in obesity(Reference Kalupahana, Moustaid-Moussa and Claycombe8). Older people also commonly exhibit low-grade chronic inflammation that may contribute to frailty, depending on the plasma levels of factors such as tumor necrosis factor (TNF) and interleukin (IL)-6.
Table 1 Features of acute, chronic and low-grade chronic inflammation

Common to both acute and chronic inflammation is that they have an afferent phase, in which the presence of a trigger is sensed by some types of cells, and an efferent phase, in which an inflammatory response is generated to eliminate the perceived hostile intruder (i.e. the source of the trigger). Irrespective of the type of inflammation, the response involves four major events. The first event is increased blood supply to the site of inflammation. The second event is increased capillary permeability caused by retraction of endothelial cells. This permits larger molecules, not normally capable of traversing the endothelium, to do so and thus delivers some soluble mediators to the site of inflammation. The third event is leucocyte migration from the capillaries into the surrounding tissue (Fig. 2). This is promoted by the release of chemoattractants from the site of inflammation and by the up-regulation of adhesion molecules on the endothelium. Once in the tissue, the leucocytes move to the site of inflammation. The fourth event is the release of mediators from leucocytes at the site of inflammation. These may include lipid mediators (e.g. prostaglandins (PG), leukotrienes; see Table 2), peptide mediators (e.g. cytokines, chemokines; see Table 3), reactive oxygen species (e.g. superoxide), amino acid derivatives (e.g. histamine) and enzymes (e.g. matrix proteases), depending upon the cell type involved, the nature of the inflammatory trigger, the anatomical site involved and the stage during the inflammatory response. Several of the mediators involved act to amplify the inflammatory process by acting, for example, as chemoattractants. Some of the inflammatory mediators escape the inflammatory site into the circulation and from there, they can exert systemic effects (Fig. 2). For example, the cytokine IL-6 released into the bloodstream from an inflammatory locus induces hepatic synthesis of the acute-phase protein C-reactive protein (CRP), while the cytokine TNF elicits metabolic effects within skeletal muscle, adipose tissue and bone. Thus, inflammation at one site, such as within the intestinal mucosa, can cause inflammation-driven changes at distal sites, such as in bone.

Fig. 2 An overview of inflammation. C, complement; CRP, C-reactive protein; SAA, serum amyloid A; TNFR, TNF receptor; ra, receptor antagonist; PAI, plasminogen activator inhibitor; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; CCL, chemokine (C–C motif) ligand; CD62E, E-selectin endothelial leukocyte adhesion molecule 1; CD62P, P-selectin endothelial leukocyte adhesion molecule 1; CXCL, chemokine (C–X–C motif) ligand; tPA, tissue plasminogen activator; vWF, von Willebrand factor; IFN, interferon; PLA, phospholipase A; ROS, reactive oxygen species.
Table 2 Lipid mediators associated with inflammation*

COX, cyclo-oxygenase; TX, thromboxane; HETE, hydroxyeicosatetraenoic acid; LOX, lipoxygenase; HPETE, hydroperoxyeicosatetraenoic acid; cys, cysteinyl; LT, leukotriene; LX, lipoxin; Rv, resolvin; P, protectin; NP, neuroprotectin; MaR, maresin; PAF, platelet-activating factor; Lyso-PA, lysophosphatidic acid; DP, EP, FP, IP, TP, prostanoid receptors; BLT, leukotriene B4 receptors; OXE, oxoeicosanoid receptor; D4, E4, Leukotriene receptors; FPR2, formyl peptide receptor 2; ALX, oncogenic receptor tyrosine kinase; CB, cannabinoid receptor; ChemR23, chemokine receptor-like 1 (also known as Chemerin receptor 23); RvD, resolving D; GPR32, G protein-coupled receptor 32; LP, lysophospholipid; S1P, sphingosine-1-phosphate.
* Note that the listing is not exhaustive.
Table 3 Soluble peptides associated with inflammation*

ra, receptor antagonist; Th, helper T; NK, natural killer; IL-18Ra, IL-18 receptor alpha; IL-1Rrp, IL1 receptor-related protein; IL-18Rb, IL-18 receptor beta; IL-18RAP, IL-18 receptor accessory protein; IFN, interferon; APC, antigen-presenting cells; R, receptor; MIP, macrophage inhibitory protein; CXCL, chemokine (C–X–C motif) ligand; GRO, growth-regulated protein; CXCR, chemokine (C–X–C motif) receptor; IP-10, interferon γ-induced protein 10; MCP, monocyte chemoattractant peptide; CCL, chemokine (C–C motif) ligand; CCR, chemokine (C–C motif) receptor; RANTES, regulated on activation, normal T expressed and secreted; TLR, Toll-like receptor; tPA, tissue plasminogen activator; VCAM, vascular cell adhesion molecule; ICAM, intercellular adhesion molecule; sPLA2, soluble phospholipase A2; PAF, platelet-activating factor; GP130, glycoprotein 130; FPR, formyl peptide receptor; CD, cluster of differentiation; SR-BI, scavenger receptor class B member 1; P2X7, P2X purinoceptor 7; uPA, urokinase receptor; LFA-1, lymphocyte function-associated antigen 1; MAC-1, macrophage-1 antigen; PSGL-1, P-selectin glycoprotein ligand 1; ESL-1, E-selectin ligand 1; LRP1, Low density lipoprotein receptor-related protein 1; Fc1R1, high-affinity IgE receptor 1; Fc1R2, high-affinity IgE receptor 2.
* Note that the listing is not exhaustive.
The need to identify biomarkers of inflammation
Inflammation has long been recognised to play a key role in the pathology of a number of human diseases, including RA, psoriasis, Crohn's disease, allergic asthma and atopic dermatitis, being centrally involved in the tissue damage that is seen. These diseases with a well-recognised inflammatory component are usually treated with general or specific anti-inflammatory pharmaceuticals with the aim of inducing a clinical benefit. More recently, it has become clear that metabolic diseases such as atherosclerosis, type 2 diabetes and obesity have an inflammatory component(Reference Calder, Ahluwalia and Brouns6, Reference Glass and Witztum9–Reference Ross11), albeit low-grade; however, the extent to which this is causal in the pathology is not yet clear. These diseases are not usually treated with anti-inflammatory pharmaceuticals. It is considered that many dietary factors can influence various aspects of inflammation(Reference Calder, Albers and Antoine1, Reference Calder, Ahluwalia and Brouns6); thus, nutrition may play a role in predisposing to conditions that have an inflammatory component and altered nutrition may be useful in the prevention or treatment of such conditions(Reference Calder, Albers and Antoine1, Reference Calder, Ahluwalia and Brouns6).
In order to elucidate the role of diet or its component foods, nutrients and non-nutrients in inflammation, either as a predisposing, preventative or therapeutic factor, there is a need to be able to monitor the inflammatory process in the subjects being studied. This requires the identification of biomarkers of inflammation. A biomarker is defined as ‘a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to an intervention’(12). However, clear recommendations on which biomarkers to use and how to interpret patterns of biomarkers and changes in biomarkers in the context of inflammation are lacking. The Institute of Medicine recently presented a general concept for identification of biomarkers and surrogate endpoints in chronic diseases(13). The Institute of Medicine recommended basing the evaluation process for biomarkers on three steps: (1) analytical validation of the biomarker (reference range, accuracy, limits of detection, reproducibility, etc.); (2) qualification of the available evidence on an association between the biomarker and disease states including data showing the effects of an intervention on both the biomarker and clinical outcomes; (3) use of a utilisation step that primarily includes the determination of whether analytical validation and qualification conducted provide sufficient support for the proposed use of the biomarker(13). The overall aim of this review is to identify robust and predictive markers, or patterns or clusters of markers, that can be used to assess inflammation in human nutrition studies in the general population and to create a consensus for grading these biomarkers.
How were the biomarkers chosen?
In order to select likely biomarkers of relevance to nutrition studies, it was decided to first identify general and disease-specific markers of inflammation within the circulation (i.e. soluble within plasma/serum or as circulating cells) and within tissue sites. These markers were identified by considering a range of conditions with a recognised inflammatory component elicited within different body compartments (i.e. adipose tissue, brain, gastrointestinal tract, vascular wall, joints, airways or skin). The objective of considering such a wide range of pathological conditions was to identify those markers that are common to inflammation, irrespective of its causative trigger. The following four general families of inflammatory disease are discussed herein: metabolic conditions (obesity, type 2 diabetes and cardiovascular disease (CVD)), immune-mediated inflammatory diseases (RA, Crohn's disease and psoriasis), allergic diseases (asthma and atopic dermatitis) and a prototypical neurodegenerative disease (Alzheimer's disease). These diseases each have different molecular triggers, which are mostly exogenous to the host individual but are endogenous in some cases. For each disease, Table 4 lists the plasma/serum (hereafter termed soluble), blood cell and tissue markers of inflammation commonly reported in the literature related to that disease; the listing is not exhaustive.
Table 4 Markers associated with inflammation in specific diseases

Th, helper T; IFN, interferon; VEGF, vascular endothelial growth factor; CXCL, chemokine (C–X–C motif) ligand; GRP, glucose-related protein; PKR, protein kinase R; SMAD, Intracellular proteins that transduce extracellular signals from TGF beta ligands; CPN, chaperone; MAPK, mitogen-activated protein kinase; LT, leukotriene; CRP, C-reactive protein; SAA, serum amyloid A; ra, receptor antagonist; R, receptor; CCL, chemokine (C–C motif) ligand; vWF, von Willebrand factor; VCAM, vascular cell adhesion molecule; ICAM, intercellular adhesion molecule; PAI, plasminogen activator inhibitor; sPLA2, soluble phospholipase A2; C, complement.
Although inflammation-induced tissue damage occurs in an organ-specific manner (adipose tissue, vascular wall, joints, skin, gut, airways and brain) in the different diseases or conditions, there is some commonality among the responses and markers seen in the different organs (summarised in Tables 5 and 6 for soluble markers and blood cellular markers, respectively). Indeed, although triggers, localisation and resulting clinical symptoms are different, many of the processes, cells and molecules involved in the actual inflammatory response are remarkably similar. Most, if not all, of the chronic inflammatory diseases considered here are characterised by an increase in the number of leucocytes in the bloodstream and by overproduction and appearance in the bloodstream of increased concentrations of inflammatory cytokines (TNF, IL-1β, IL-6 and interferon-γ) and chemokines (IL-8 and monocyte chemoattractant protein (MCP)-1). Elevated levels of these mediators act to amplify the inflammatory process (e.g. by attracting further inflammatory cells to the site), contribute to tissue destruction, elicit systemic effects (e.g. hepatic CRP synthesis) and, in many cases, actually cause the clinical symptoms observed (Fig. 2; Table 4). Thus, a consideration of a wide range of inflammatory diseases affecting distinct sites within the body has enabled the identification of a set of ‘common’ or ‘general’ markers of inflammation that can be measured in the bloodstream (Tables 5 and 6). The selection of soluble markers of inflammation has focused on peptides and proteins; lipid mediators have not been considered further because of their instability and relative difficulty of measurement. For most of the soluble markers identified (Table 5), there is no commonly accepted normal range of concentrations. In contrast, for the blood cellular markers of inflammation (Table 6), there is a generally accepted normal range and some of these markers are used clinically.
Table 5 Soluble markers of inflammation and their associated modifying factors

R, receptor; IFN, interferon; CCL, chemokine (C–C motif) ligand; CRP, C-reactive protein; SAA, serum amyloid A; C, complement; ra, receptor antagonist; VCAM, vascular cell adhesion molecule; ICAM, intercellular adhesion molecule; ↑ , positive association; ↓ , negative association; ↔ , no clear association.
* These values are based on groups of healthy subjects in studies published in scientific literature. These are therefore indicative of what could be expected in such populations.
† Values >10 mg/l considered to be indicative of inflammation beyond ‘low grade’.
Table 6 Blood cellular markers of inflammation and their modifying factors

Up arrow, Positive association; Down arrow, Negative association; Double arrow, No clear association.
* Based on data reported in MacLennon & Drayson(Reference MacLennon, Drayson, Tuddenham, Hoffbrand and Lewis336).
In addition to the general, non-specific markers of inflammation identified above, there are a number of inflammatory markers that are more disease specific (Table 4). Sometimes these markers are present at the site of the inflammatory locus, such as thymic stromal lymphopoietin in skin lesions in atopic dermatitis. On other occasions, they may appear in the circulation (e.g. rheumatoid factor in RA). Because the aim of this evaluation of inflammatory markers is to identify those to be assessed in nutrition studies in the general population, disease-specific markers are not considered to be of relevance to this aim. Likewise, assessment of general, non-specific inflammatory markers at sites of disease activity (e.g. cytokines in synovial fluid in RA, in bronchoalveolar lavage fluid in asthma, in skin lesions in psoriasis or in gut mucosa in Crohn's disease) was not considered relevant to the non-disease setting. For these reasons, inflammatory markers that are disease specific and assessment of inflammatory markers in tissues or lesions are not further considered here. Thus, this review is restricted to the consideration of inflammatory markers in the blood, either soluble markers in plasma/serum (Table 5) or circulating cells (Table 6).
Key messages
(1) Acute inflammation is a normal physiological response critical to maintain homeostatic control, but when it becomes chronic, inflammation contributes to the pathophysiology of a range of diseases.
(2) Common soluble markers of inflammation include cytokines, chemokines, lipids, acute-phase proteins and adhesion molecules.
(3) Common blood cellular markers of inflammation include various subclasses of leucocytes. The same markers are involved in both acute and chronic inflammatory processes, meaning that the markers themselves, in isolation, cannot be used to distinguish the two processes.
Modifiers of inflammatory markers
There is a considerable between-individual variation in the numbers of leucocytes and of leucocyte subclasses and in the concentrations of the soluble markers of inflammation. There is no generally accepted ‘normal range’ for most of these markers, although there are clinically accepted normal ranges for total leucocytes and the leucocyte subclasses (Table 6). A number of modifiers, other than the presence of an inflammatory trigger, have been shown to influence the concentrations of these markers; these modifiers include age, body fatness, physical (in)activity, sex, genetics, smoking, gut microbiota composition, diet, use of medications and other factors such as emotional stress, pollution, viral infection and sleep behaviour (Fig. 3; Tables 5 and 6). These modifiers should all be considered when designing a study aimed at measuring inflammatory biomarkers.
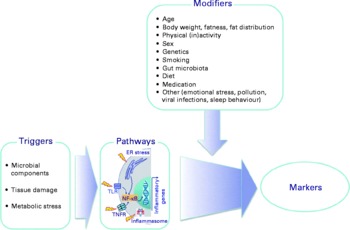
Fig. 3 Factors that influence the status of soluble and cellular markers of inflammation in the bloodstream. Triggers are those factors that can directly initiate an inflammatory response. Modifiers are endogenous and exogenous factors that may increase or decrease the response induced by a trigger. ER, endoplasmic reticulum; TLR, Toll-like receptor; TNFR, TNF receptor.
Influence of age
A certain degree of inflammation may be part of adaptability and homeostasis in older people, and it has been demonstrated that their ability to mount a vigorous inflammatory response is associated with enhanced survival(Reference Ferrucci, Corsi and Lauretani14). On the other hand, inflammation is considered to play an important role in the loss of lean body mass and in progression towards frailty in older persons. Studies investigating a possible effect of age on inflammatory markers have generally adopted one of two approaches. The first has been to look for an association between age and the concentrations of markers of interest, whereas the second has been to compare marker concentrations between two or more groups of individuals of different ages. Studies have generally stratified populations and set arbitrary thresholds varying from ages >60, 65, 70 to ≥ 75 years to identify aged individuals. These studies, for the most part, have suggested that ageing is accompanied by a low-grade chronic inflammatory state, clearly shown by two- to fourfold higher concentrations of serum levels of several inflammatory markers such as cytokines and acute-phase proteins in older persons(Reference Krabbe, Pedersen and Bruunsgaard15). Several studies have reported age-associated increases in total leucocyte numbers(Reference Otsuka, Tamakoshi and Wada16, Reference Rana, Boekholdt and Ridker17) and in soluble concentrations of soluble pro-inflammatory cytokines such as IL-1(Reference Fagiolo, Cossarizza and Scala18, Reference Pietschmann, Gollob and Brosch19), IL-6(Reference Ferrucci, Corsi and Lauretani14, Reference Fagiolo, Cossarizza and Scala18, Reference Hager, Machein and Krieger20, Reference Roubenoff, Harris and Abad21) and TNF(Reference Paolisso, Rizzo and Mazziotti22, Reference Bruunsgaard, Andersen-Ranberg and Jeune23), although this has not been seen in all studies(Reference Roubenoff, Harris and Abad21, Reference Shurin, Yurkovetsky and Chatta24). Others have reported higher concentrations of IL-1 receptor antagonist(Reference Ferrucci, Corsi and Lauretani14, Reference Roubenoff, Harris and Abad21, Reference Catania, Airaghi and Motta25) and of TNF-receptors(Reference Shurin, Yurkovetsky and Chatta24, Reference Catania, Airaghi and Motta25) with increased age. In line with these findings, many studies have reported higher concentrations of downstream markers such as acute-phase proteins like CRP(Reference Ferrucci, Corsi and Lauretani14, Reference Yasuma, Nakamura and Nishiguchi26), α1-acid glycoprotein(Reference Akbay, Yetkin and Ersoy27) and fibrinogen(Reference Ferrucci, Corsi and Lauretani14, Reference Hager, Machein and Krieger20, Reference Aliberti, Proietta and Pulignano28). Fewer studies have addressed the relationship of ageing with chemokines and adhesion molecules, although in some studies, vascular cell adhesion molecule (VCAM)-1(Reference Forsey, Thompson and Ernerudh29, Reference Richter, Rassoul and Purschwitz30) and intercellular adhesion molecule (ICAM)-1(Reference Richter, Rassoul and Purschwitz30) have been shown to have higher concentrations in older persons while E-selectin was not(Reference Richter, Rassoul and Purschwitz30). Similarly, there is some evidence that concentrations of MCP-1(Reference Seidler, Zimmermann and Bartneck31), plasminogen activation inhibitor-1(Reference von Eyben, Mouritsen and Holm32) and adiponectin(Reference Koh, Hyun and Choi33, Reference Ku, Kim and Lee34) are increased with ageing.
Overall, these changes support a generally increased state of low-grade chronic inflammation in older people compared with younger adults. The more limited literature on blood cellular markers would support this contention, but indicates that numbers and proportions of leucocytes in the older people differ considerably depending on the state of health of the individual(Reference Remarque and Pawelec35). Although older individuals have been reported to have increased total leucocytes, the increase in neutrophils, in particular, has been related to reduced survival in longitudinal studies, whereas total lymphocyte numbers are not significantly associated with survival(Reference Strindhall, Nilsson and Lofgren36). However, within the total lymphocyte population, there may be decreased numbers of B cells and increased numbers of CD8 cells in older persons, which can also have an impact on survival and which to a great extent depend on microbiological exposures(Reference Muller, Pawelec, Derhovanessian, Calder and Yaqoob37).
It is recognised that ageing represents cumulative effects of exposure to various triggers such as diet, physical (in)activity and stress, as well as micro-organisms, over the lifetime, and it is hypothesised that failure of anti-inflammatory mechanisms to neutralise such inflammatory processes triggered continuously lifelong plays a role in low-grade chronic inflammation in older people(Reference Franceschi38). Inflammatory markers in older persons may also be influenced by co-morbidity, medication use or malnutrition(Reference Ahluwalia39–Reference Jenny, Tracy and Ogg43). However, there are some indications that once co-morbidities are accounted for, inflammation is still associated with ageing(Reference Ferrucci, Corsi and Lauretani14, Reference Krabbe, Pedersen and Bruunsgaard15). A number of other factors that may affect and modulate circulating levels of inflammatory mediators alter with ageing, including body composition, physical (in)activity, sex hormone concentrations, gut microbiota, emotional stress and a poor social environment(Reference Candore, Colonna-Romano and Balistreri44–Reference Biagi, Nylund and Candela48). It is likely that in some studies, these confounders have not been considered when the effect of age on inflammation has been evaluated(Reference Candore, Colonna-Romano and Balistreri44–Reference Biagi, Nylund and Candela48). Thus, it is challenging to dissect out the effect of ageing per se on inflammatory markers in humans and essentially impossible to exclude all residual confounding concomitant modifying factors and lifelong exposures. One possibility is to examine inflammatory markers in centenarians as a model of successful ageing. Low levels of the pro-inflammatory cytokine IL-6 and high levels of anti-inflammatory cytokines such as IL-10 and transforming growth factor (TGF) have been noted in centenarians(Reference Lio, Scola and Crivello49, Reference Iannitti and Palmieri50), suggesting that these extremely old persons do not suffer the ‘inflammaging’ noted in many older persons who do not survive to 100 years of age or beyond.
Influence of body fatness and fat distribution
As stated earlier, white adipose tissue secretes cytokines and chemokines, and macrophage and T-cell infiltration is dramatically increased in the adipose tissue of obese humans(Reference Weisberg, McCann and Desai51). Obesity is associated with a state of low-grade chronic inflammation. Total blood leucocyte numbers are positively associated with obesity and the metabolic syndrome(Reference Otsuka, Tamakoshi and Wada16, Reference Rana, Boekholdt and Ridker17, Reference Lee, Shin and Kim52–Reference Skinner, Steiner and Henderson54). Differential leucocyte counts indicate that neutrophils, but not monocytes or lymphocytes, account for this predictive potential(Reference Rana, Boekholdt and Ridker17). There are higher concentrations of circulating cytokines and chemokines such as TNF(Reference Corica, Allegra and Corsonello55–Reference Ziccardi, Nappo and Giugliano57), IL-1(Reference Meier, Bobbioni and Gabay58–Reference Ueland, Kristo and Godang60), IL-6(Reference Ziccardi, Nappo and Giugliano57, Reference Bastard, Jardel and Bruckert61, Reference Yudkin, Stehouwer and Emeis62), IL-8(Reference Kim, Park and Kawada63, Reference Straczkowski, Dzienis-Straczkowska and Stepien64) and MCP-1(Reference Kim, Park and Kawada63, Reference Christiansen, Richelsen and Bruun65, Reference Schernthaner, Kopp and Kriwanek66) in obese individuals compared with normal-weight subjects. Several acute-phase proteins are elevated in overweight and obese subjects. For example, the circulating concentration of CRP is higher in obese persons, and obese subjects who lost weight showed decreased levels of CRP(Reference Tchernof, Nolan and Sites67, Reference Visser, Bouter and McQuillan68). A meta-analysis showed a strong association between Body Mass Index (BMI) and serum amyloid A concentration(Reference Zhao, He and Shi69). Elevated levels of soluble adhesion molecules, such as VCAM-1, ICAM-1, E-selectin and P-selectin, are found in obese individuals compared with normal-weight subjects(Reference Ziccardi, Nappo and Giugliano57, Reference Hanusch-Enserer, Zorn and Wojta70–Reference Zanni, Stanley and Makimura74). Leptin concentrations are higher in obese than in normal-weight subjects(Reference Corica, Allegra and Corsonello55, Reference Bastard, Jardel and Bruckert61, Reference Christiansen, Richelsen and Bruun65, Reference Maachi, Pieroni and Bruckert75), while adiponectin concentrations are lower in obese individuals(Reference Arita, Kihara and Ouchi76–Reference Yang, Lee and Funahashi78). Although these studies have focused on obesity per se, the distribution of adipose tissue appears to be important in determining inflammatory burden. Visceral adipose tissue has a greater production of inflammatory mediators such as cytokines than subcutaneous adipose tissue, although the precise contribution of each to the circulating pool is not clear(Reference Weiss79). Furthermore, other adipose tissue depots in so-called ‘ectopic sites’, such as within the liver, heart or skeletal muscle, may contribute to the production of inflammatory mediators even in the absence of obesity(Reference Greenfield and Campbell80). In this regard, the local production of inflammatory molecules by adipose tissue within the heart may be important; the amount of this tissue and its proximity to the coronary vessels could contribute to the development of coronary pathologies(Reference Rabkin81, Reference Silaghi, Piercecchi-Marti and Grino82). Although there has been considerable focus on the role of low-grade chronic inflammation in overweight, obesity and the metabolic syndrome(Reference Calder, Ahluwalia and Brouns6), it is important to recognise that underweight also appears to be associated with low-grade chronic inflammation. For example, adolescent anorexia is associated with elevated levels of some inflammatory cytokines including TNF-α(Reference Ondrak and Hackney83), and low-grade chronic inflammation is associated with low body weight in older subjects(Reference Nakajima, Yamaoka and Morita84). Several pro-inflammatory cytokines, most notably TNF-α, have a causal role in cachexia(Reference Stephens, Skipworth and Fearon85).
Influence of physical (in)activity
Physical (in)activity resulting in inadequate energy expenditure relative to intake may lead to the accumulation of visceral fat and consequently to the activation of inflammatory processes associated with this(Reference Martinez-Gomez, Eisenmann and Gomez-Martinez86). In contrast, physical activity promotes health and this may relate to the anti-inflammatory effect of regular exercise, which reduces visceral fat mass and induces an anti-inflammatory environment(Reference Walsh, Gleeson and Shephard87, Reference Walsh, Gleeson and Pyne88). Subjects engaged in regular physical activity have lower blood leucocyte numbers(Reference Johannsen, Priest and Dixit89). Plasma concentrations of IL-6 are lower in individuals who undertake regular physical activity than in those who do not(Reference Hamer90). The lower concentrations of IL-6 in the circulation will subsequently result in lower CRP levels. Several studies of large population cohorts have provided evidence for an inverse, independent dose–response relationship between the level of habitual physical activity and plasma CRP concentration in both men and women(Reference Abramson and Vaccarino91–Reference Wannamethee, Lowe and Whincup96). Moreover, CRP levels in 2120 Finnish participants were associated with obesity indices (positively) and physical activity (inversely) among both sexes(Reference Raitakari, Mansikkaniemi and Marniemi97). Cross-sectional studies have demonstrated that cardiorespiratory fitness levels are inversely associated with CRP concentrations and also the prevalence of elevated CRP concentrations(Reference Church, Barlow and Earnest98). However, conflicting findings exist and several training interventions have not produced changes in basal IL-6 or CRP levels(Reference Bautmans, Njemini and Vasseur99–Reference Martinez-Gomez, Gomez-Martinez and Ruiz106). There seems to be more consistency in patients with metabolic or coronary disease and in older individuals than in healthy or young people. In a relatively large intervention study of exercise training in cardiac rehabilitation patients, CRP concentration was reduced by 41 %, whereas CRP concentrations did not change in subjects who did not exercise(Reference Milani, Lavie and Mehra107). Rankovic et al. (Reference Rankovic, Milicic and Savic108) showed that 6 weeks of aerobic exercise can significantly reduce the inflammatory state by decreasing CRP and VCAM-1 levels in patients with stable coronary heart disease. One possible explanation could be that patients and older people have higher basal levels of inflammation before becoming physically active and physical activity is more effective in lowering the CRP levels in those with the highest initial CRP concentrations(Reference Milani, Lavie and Mehra107). Given that physical activity and obesity are often inversely related, it is not clear as to whether the anti-inflammatory health benefits of a physically active lifestyle are due to exercise per se or result from favourable changes in body composition. A systematic review addressed whether fitness or fatness has the greatest impact on inflammatory factors(Reference Hamer90) and concluded that both fatness and the lack of fitness are associated with systemic inflammatory status, although the relative contributions of both may be dependent on age, disease status and sex. Different types of exercise to attain and maintain a state of physical fitness may have different effects on inflammation(Reference Martinez-Gomez, Eisenmann and Gomez-Martinez109), although this is little studied.
Influence of sex
Few studies have examined the effect of sex on inflammatory markers and the literature presents mixed findings. Some studies have reported no differences between males and females for most inflammatory markers examined, including TNF, TNF-receptor, IL-1, MCP-1(Reference Shurin, Yurkovetsky and Chatta24) or IL-6 and ICAM-1(Reference Forsey, Thompson and Ernerudh29). However, in a study of adolescents (aged 13 to 17 years), although CRP, C3 and C4 concentrations did not differ between girls and boys, adolescent boys had lower levels of adiponectin, leptin and l-selectin than girls, whereas girls had lower levels of ICAM-1, VCAM-1 and plasminogen activation inhibitor-1 than boys(Reference Martinez-Gomez, Eisenmann and Gomez-Martinez86). Some investigators have reported lower IL-1β levels in men(Reference Marques-Vidal, Bochud and Bastardot110), whereas others(Reference Bouman, Schipper and Heineman111) have found that men have higher levels of IL-1β-secreting monocytes than women. Importantly, few studies have reported on sex effects after adjusting for variables that can affect inflammatory markers, such as body fatness, fat distribution and physical (in)activity. Marques-Vidal(Reference Marques-Vidal, Bochud and Bastardot110) showed in a Swiss population-based sample that males have higher levels of IL-6 and TNF and slightly but significantly lower levels of CRP when compared with females in crude analysis and after adjusting for age, BMI, smoking status and leisure-time physical activity in multivariate analysis. There is a need for further population-based studies in healthy adults to examine sex differences in inflammatory markers taking into account potential important confounders.
Influence of genetics
Both epigenetic and genomic variations influence the extent of the response of an inflammatory marker to a given trigger. Epigenetics refers to chemical modifications to chromatin or DNA that influence gene expression but are not encoded in the DNA sequence itself. The two major epigenetic mechanisms are the post-translational modification, often acetylation, of histone proteins in chromatin and the methylation of DNA itself. DNA methylation occurs at CpG sites of the gene sequence. Epigenetic variations appear to play an important role in determining the risk of inflammatory disorders(Reference Durham, Chou and Kirkham112–Reference Wierda, Geutskens and Jukema114). Differences in the methylation status of CpG sites have been observed in key inflammatory response genes and influence the expression of those genes(Reference Wilson115). This is an area in which there are likely to be major advances in the next few years. It is important to note that epigenetic modifications are not fixed but may be modified by a number of different exposures, including stress hormones, oxidative stress, pollution and diet. This may partly explain the ability of those factors to act as modifiers of the inflammatory response.
SNP are variations in the DNA sequence at a single nucleotide that occur in at least 1 % of the population. They can occur in both coding and non-coding regions of the genome. It is estimated that the human genome contains 3 million SNP. Genotype is a description of the allele pair present at a given site in the genome, and typically there will be three possible genotypes (e.g. AA, AG and GG). There are variations in allele frequencies and genotypes among different human populations because certain alleles and genotypes may have offered an evolutionary advantage in one geographical region and not in another. SNP within a coding sequence do not necessarily change the amino acid sequence of the protein that is produced, because of the degeneracy of the genetic code. SNP that are not in protein-coding regions may affect transcription factor binding or gene splicing or the sequence of non-coding RNA and, as a result, the gene expression, and hence the level of the encoded protein may differ among individuals with different genotypes. It is likely that SNP occur in regions of the genome associated with each of the inflammatory proteins being considered here (e.g. cytokines, chemokines, adhesion molecules, acute-phase proteins), encoding enzymes responsible for the synthesis of non-peptide inflammatory mediators (e.g. in cyclo-oxygenase and lipoxygenase enzymes, NADPH oxidase and inducible NO synthase) and encoding regulatory proteins such as transcription factors. In addition, such genetic variations underlie the huge number of possible variants in the human leucocyte antigens involved in antigen presentation. This variation in possible human leucocyte antigens allows an individual to have the potential to deal with an enormous number of possible antigens that may be encountered. However, some human leucocyte antigens variants have strong associations with inflammatory diseases, such as human leucocyte antigens-DR4 with RA. A number of SNP are of functional significance, meaning that individuals with different genotypes will produce different levels of a particular inflammatory mediator in response to a given stimulus or trigger than other individuals(Reference Fishman, Faulds and Jeffery116–Reference Santtila, Savinainen and Hurme119). Thus, genotype can influence the plasma concentration of inflammatory markers(Reference Fishman, Faulds and Jeffery116, Reference Stuber, Petersen and Bokelmann120, Reference Kelberman, Fife and Rockman121). For example, the G to C polymorphism at the − 174 position of the IL-6 gene has been reported to significantly influence plasma IL-6 concentrations: mean concentrations (pg/ml) after adjusting for age, BMI, sex and smoking were 2·74 for GG homozygotes, 2·64 for heterozygotes and 1·63 for CC homozygotes(Reference Fishman, Faulds and Jeffery116). Similarly, the polymorphism at the +252 position of the TNF-β gene significantly affected plasma TNF concentrations in patients with sepsis: median concentrations (pg/ml) were approximately 200 for B1 homozygotes, 250 for heterozygotes and 600 for B2 homozygotes(Reference Stuber, Petersen and Bokelmann120). The genotype of certain inflammatory genes has been associated with the risk or severity of outcome in inflammatory disease(Reference Wilson, Gordon and di Giovine122, Reference Wilson, di Giovine and Duff123), infectious disease(Reference Wilson, di Giovine and Duff123, Reference Read, Camp and di Giovine124) and cancer(Reference Wilson, di Giovine and Duff123, Reference Howell, Turner and Collins125).
Ethnicity is related to genetic variation. The concentrations of some inflammatory markers have been shown to differ among ethnic groups(Reference Miller and Cappuccio126) and the prevalence of many of the common inflammatory diseases differs according to ethnicity. However, in addition to genetics, ethnic groups often differ in a number of the other modifying factors, which may contribute to the observed differences in inflammatory markers. In a systematic review and meta-analysis involving 221 287 people from eighty-nine studies, median CRP concentrations were 2·60 (95 % CI 2·27, 2·96), 2·51 (95 % CI 1·18, 2·86), 2·34 (95 % CI 1·99, 2·80), 2·10 (95 % CI 1·77, 2·30) and 1·01 (95 % CI 0·88, 1·18) mg/l in African Americans, Hispanics, South Asians, Caucasians and East Asians, respectively(Reference Shah, Newcombe and Smeeth127). The differences among groups were retained after adjusting for age and BMI.
Influence of smoking
Cigarette smoke contains a variety of oxidant molecules in high amounts, inducing a strong oxidative stress in the smoker. In turn, oxidative stress can induce an inflammatory response(Reference Calder, Albers and Antoine1). Indeed, it has been established in several population-based studies that smoking is associated with a low-grade chronic inflammatory state, as shown by higher blood leucocyte numbers(Reference Ernst128, Reference Holz, Thorand and Doring129) and higher circulating concentrations of acute-phase proteins and cytokines. Several studies have reported elevated circulating concentrations of pro-inflammatory cytokines such as IL-6(Reference Bermudez, Rifai and Buring130–Reference Woodward, Rumley and Tunstall-Pedoe132) and TNF(Reference Petrescu, Voican and Silosi133, Reference Wirtz, von and Kunz-Ebrecht134) in smokers compared with non-smokers. A number of studies have reported that CRP and fibrinogen concentrations are higher in smokers than in non-smokers(Reference Bazzano, He and Muntner135–Reference Yarnell, Sweetnam and Rumley140). CRP and fibrinogen concentrations are found to correlate significantly with the years of smoking, even after smoking cessation. In the Multinational MONItoring of trends and determinants in CArdiovascular disease (MONICA) study, for example, CRP concentrations were still elevated 10 years after quitting smoking(Reference Frohlich, Sund and Lowel136). Some adhesion molecules are also increased in the serum of smokers compared with non-smokers, including ICAM-1, P-selectin and E-selectin(Reference Bermudez, Rifai and Buring130, Reference Bergmann, Siekmeier and Mix141). Taken as a whole, these are strong data supporting that smoking is associated with low-grade chronic inflammation. Fewer studies have investigated the impact of long-term passive smoking in non-smokers on inflammatory markers; those studies that have been published present inconsistent findings. In a recent study on 200 non-smoking US truck industry workers, no statistically significant association was found between chronic exposure to second-hand smoke and systemic inflammatory markers such as IL-6, CRP or ICAM-1(Reference Chiu, Spiegelman and Dockery142). Furthermore, in 7600 never-smoking adults included in National Health and Nutrition Examination Survey (NHANES) III, no significant association was found between CRP and second-hand smoke exposure, measured as serum cotinine level; however, fibrinogen was significantly higher in subjects with detectable but low cotinine levels(Reference Venn and Britton143). However, in another study conducted on 5000 older British non-smoking subjects, serum cotinine level was positively associated with CRP, fibrinogen, von Willebrand factor and tissue plasminogen activator(Reference Jefferis, Lowe and Welsh144).
Influence of gut microbiota
Humans co-evolved with gut microbes, which represent more than 3 million genes from several hundreds of bacterial species. There is evidence that the nature of the gut microbiota influences inflammatory processes, particularly within the gastrointestinal tract, but perhaps also systemically(Reference Chow, Tang and Mazmanian145–Reference Maslowski and Mackay149). Thus, in animal models of IBD, disease initiation strongly depends on the presence of commensal bacteria and, conversely, in germ-free animals, IBD hardly develops(Reference Jiang, Kushnir and Thurnheer150, Reference Seksik151). In support of this, human studies have revealed that dysbiosis (gut microbiota alteration) is a common feature in patients with IBD in which the overall bacterial diversity is reduced and species belonging to the phyla Actinobacteria and Proteobacteria are over-represented(Reference Seksik151–Reference Walker, Sanderson and Churcher154) while the phylum Firmicutes is reduced, especially Faecalibacterium prausnitzii, which has anti-inflammatory properties(Reference Sokol, Pigneur and Watterlot155, Reference Sokol, Seksik and Furet156). However, it is still unclear whether these changes are responsible for causing disease or are a result of other changes in the gut environment that result from inflammatory reactions and mucosal tissue damage. Nonetheless, some recent studies have reported positive effects of using probiotics, prebiotics and their combination (termed synbiotics) in IBD by a restoration of a healthy gut microbiota(Reference Claes, De Keersmaecker and Vanderleyden157, Reference Gareau, Sherman and Walker158). This suggests that there is potential for treating IBD through microbiota manipulation and modifications of interactions with the gut immune system. These findings provide a rationale for microbial characterisation in many other inflammatory diseases, such as psoriasis and allergic asthma. As in IBD, specific alterations of skin and airway microbiota are observed in patients with inflammatory diseases manifested at these sites(Reference Cogen, Nizet and Gallo159–Reference Paulino, Tseng and Blaser164). More recently, gut microbiota in atopic dermatitis and in obese people has also been carefully addressed and a specific profile was found, indicating that gut commensals may also exert remote effects(Reference Chow, Tang and Mazmanian145, Reference Delzenne and Cani165, Reference Penders, Stobberingh and van den Brandt166). However, the precise causal role and the mechanisms involved remain to be established. Recent findings have also suggested that gut microbiota is a factor in metabolic diseases, as reviewed recently(Reference Cani, Osto and Geurts167, Reference Clarke, Murphy and Nilaweera168). It has been demonstrated that the gut microbial profile is specific to the metabolic phenotype in mice and humans (i.e. obese v. lean and diabetic v. healthy). However, a clear demonstration connecting intestinal microbiota to metabolic diseases remained absent until recently. Indeed, Serino et al. (Reference Serino, Luche and Gres169) showed that diabetic mice fed a high-fat diet and that developed diabetes display a specific alteration of gut microbiota with increased gut paracellular permeability leading to higher endotoxaemia and subsequent systemic low-grade inflammation and metabolic syndrome. Conversely, mice under the same feeding protocol that did not develop disease had no gut microbiota alteration(Reference Serino, Luche and Gres169, Reference Cani, Bibiloni and Knauf170). The same specific gut microbiota alterations have also been reported in obese v. lean human subjects (reduced global diversity and relative increase in the ratio of Bacteroidetes:Firmicutes). F. prausnitzii was reduced in obese diabetic subjects compared with lean controls and was negatively correlated with serum levels of circulating inflammatory markers such as CRP and IL-6 in these subjects(Reference Furet, Kong and Tap171). These changes affect the metabolic potential of the gut microbiota and support an increased capacity to harvest energy from the diet in obese people(Reference Turnbaugh, Ley and Mahowald172–Reference Jumpertz, Le and Turnbaugh174). These features of obesity are transmissible as faecal material transfer from obese human adults to germ-free mice resulted in higher total body fat accumulation over time in contrast to lean human adult faecal material transfer(Reference Turnbaugh, Ley and Mahowald172–Reference Jumpertz, Le and Turnbaugh174).
Influence of diet
The influence of diet on soluble markers of inflammation in different contexts has been described in detail elsewhere(Reference Calder, Albers and Antoine1, Reference Calder, Ahluwalia and Brouns6) and will not be considered further here.
Influence of medication
It is obvious that whether specific or non-specific to particular disease processes and pathways or to a particular disease, pharmacologic treatment of inflammation is bound to greatly influence biomarker levels. A classic example is the influence of corticosteroids in autoimmune, allergic or mucosal inflammatory conditions; corticosteroids are powerful modifiers of cytokines and growth factors that influence other biomarkers such as CRP levels in RA and eosinophil levels in asthma. Indeed, such biomarkers are valuable in monitoring therapeutic responses and adherence to treatment. Selective treatments such as targeted biologics (e.g. anti-TNF therapy in inflammatory joint and bowel diseases) can also influence biomarkers and, again, these may be used to monitor therapy. It is, therefore, of utmost importance when using biomarkers for clinical decision making that any concurrent therapy that may influence their level or expression is taken into account.
Influence of other modifiers
In addition to the modifiers already considered, inflammatory biomarkers may be influenced by emotional stress(Reference Gouin, Hantsoo and Kiecolt-Glaser175–Reference Heffner177), exposure to pollution(Reference van Eeden, Yeung and Quinlam178) and sleep behaviour(Reference Lorton, Lubahn and Estus179) among other factors.
Summary
Among non-diseased individuals, the concentrations of the soluble markers and the numbers of some of the cellular markers of inflammation are altered with increasing age, body fatness and physical (in)activity, and are modified by genetics, smoking status, gut microbiota and medication use among other factors (Fig. 3). Typically, a number of the panel of inflammatory markers identified are altered in the same direction in response to a given modifying factor (Tables 5 and 6). Thus, there is little specificity in the inflammatory markers whose concentrations are changed during inflammatory states or which are sensitive to modifying factors. Thus, it is not possible to identify a single marker, or even a small number of markers, to define inflammation and to use as a target or targets in studies evaluating nutritional interventions. Therefore, it is likely to be important to identify patterns or clusters of markers that provide an inflammatory signature; it would seem likely that these patterns or clusters would include at least some of the individual markers considered here so far.
Key messages
(1) There are a number of endogenous and exogenous modifiers that influence the levels of inflammatory markers resulting in significant between-individual variation.
(2) These modifiers include age, body fatness, physical (in)activity, sex, genetics, smoking habits, diet, the composition of the gut microbiota and the use of certain medications.
(3) These modifiers should be recognised, and, where possible, controlled for, when nutrition studies are designed.
Challenge models
Why challenge models are relevant
The forgoing discussion has focused on individual markers of inflammation typically measured in the blood of individuals in the resting (e.g. fasting) state. Such measurements may differentiate between those individuals with an overt inflammatory disease and those without, and may also indicate the effects of influencing factors such as age, body fatness, physical (in)activity, genetics, smoking habit and the composition of the gut microbiota on chronic inflammatory tone. However, such measurements provide little information about an individual's (acute) inflammatory response to a challenge. It has been suggested that dynamic responses to challenges provide a more sensitive and meaningful indication of health(Reference Huber, Knottnerus and Green180, Reference van Ommen, Keijer and Heil181). More specifically, the dynamic inflammatory responses to a physiological challenge may provide a better (more relevant, sensitive, better interpretable) indicator of the impact of nutrition on inflammatory homeostasis and thus on the control of (acute) inflammatory responsiveness. Currently, relatively little is known about inflammatory markers after well-controlled acute challenges and whether these can be used to assess the effect of nutritional interventions on acute and/or chronic inflammation. It is also not known what exactly constitutes an optimal inflammatory response to a challenge. This will need to be established by comparing standardised responses with challenges in different (sub)populations and by exploring their predictive value in prospective cohort studies. Despite these current gaps in knowledge, assessment of inflammatory resilience after experimental pro-inflammatory challenges is increasingly being studied. This section describes the inflammatory challenges that have been used to date and makes conclusions about their utility with regard to human studies involving nutritional interventions. A range of challenges that induce an acute inflammation was identified from the literature (Table 7). These challenges can be clustered according to the dominant stress triggering the inflammatory response: metabolic stress or metabolic overload (oral glucose load and oral lipid load); infection stress (injection of pro-inflammatory stimuli such as lipopolysaccharide (LPS), TNF or IL-6; early response to (adjuvants in) vaccines; or delayed-type and contact hypersensitivity responses); or tissue damage (acute exercise or exposure to UV radiation). Typically, the same inflammatory mediators as described previously (e.g. TNF, IL-6, CRP) have been used to follow the dynamic inflammatory response to the different challenges used (Table 7).
Table 7 Challenge models that may be used to study an inflammatory response and their features

CRP, C-reactive protein; T2D, type 2 diabetes; LPS, lipopolysaccharide; CCL, chemokine (C–C motif) ligand; DTH, delayed-type hypersensitivity; ↑ , positive association; ↓ , negative association; ↔ , no clear association.
* Times indicated in parentheses are the approximate times of the maximal plasma concentration of the marker.
Metabolic stress
Oral glucose challenge
The oral glucose tolerance test (OGTT) is commonly used to test for insulin resistance and diabetes. After an overnight fast, a standardised drink containing glucose is given and subsequent blood glucose and sometimes blood insulin responses are assessed. There are some variations in the details of the protocol used(Reference Antuna-Puente, Disse and Rabasa-Lhoret182, Reference van Leeuwen, Louwerse and Opmeer183), but 75 g (1255 kJ (300 kcal)) of glucose are typically given as a standardised drink in accordance with the recommendations of the World Health Organization (WHO)(184). The oral glucose load induces a transient inflammatory response, which has been assessed in several studies as levels of circulating cytokines. Levels of CRP and IL-6 consistently increase, peaking after 1 and 2–4 h, respectively(Reference O'Keefe and Bell185–Reference Kempf, Rose and Herder190). Responses of TNF seem to be more variable. Several studies have described increases in markers of vascular inflammation such as ICAM-1(Reference Frood191), VCAM-1 and E-selectin, peaking about 2 h(Reference Derosa, D'Angelo and Salvadeo186). Most markers normalise 3–4 h after the OGTT, but responses tend to be extended and/or stronger in obese, insulin-insensitive and type 2 diabetic patients(Reference Kempf, Rose and Herder190).
Oral fat challenge
Testing responsiveness to an oral challenge with fat is sometimes referred to as the oral fat tolerance test. However, unlike the test (OFTT), this is not an established diagnostic test with a standardised WHO-recommended protocol. Quality (fatty acid composition) and quantity of the fat have differed between studies. The fat is provided as a bolus (e.g. 200–350 ml whipping cream) in some studies, whereas it is provided as a high-fat meal in other studies (e.g. 50–75 g fat as part of a meal). Most studies have focused on the effects of the OFTT on fatty acid handling, but the effects on markers of (vascular) inflammation have also been described. Postprandial lipaemia as induced by the OFTT is associated with a transient increase in markers of (vascular) inflammation, but the size and kinetics of these responses are quite variable between studies, undoubtedly due in part to the different protocols used. It has been suggested that the responses differ between healthy individuals and the obese as well as those with type 2 diabetes(Reference Ziccardi, Nappo and Giugliano57, Reference Esposito, Pontillo and Di Palo192) and are sensitive to modulation by pharmaceutical, lifestyle and dietary interventions(Reference O'Keefe and Bell185–Reference Kempf, Rose and Herder190, Reference Derosa, Ferrari and D'Angelo193). The response has typically been followed in terms of plasma concentrations of cytokines, such as TNF, IL-1β, IL-6 and IL-8, soluble adhesion molecules, such as VCAM-1 and ICAM-1, and, in some cases, circulating levels of endotoxin over the period up to 3–6 h following the fat challenge.
Infection stress
Challenge with endotoxin or inflammatory mediators
Inflammatory cascades following intravenous administration of endotoxin (bacterial endotoxin; LPS), IL-6, TNF or hydrocortisone have been studied in quite some detail, mainly in the context of understanding septic shock responses. These studies varied widely with respect to subjects, dosing and sampling regimens used, but similar response patterns emerge (Fig. 4) (Reference Taudorf, Krabbe and Berg194–Reference Krogh-Madsen, Moller and Dela198). Following intravenous administration of LPS, there is an initial increase in TNF and IL-1 peaking about 2 h, a rise in IL-6 reaching its maximum about 3 h, followed by increases in IL-8 and anti-inflammatory factors such as TNF-receptor, IL-1 receptor antagonist and IL-10 that plateau between 3 and 6 h. However, it should be noted that these are very invasive challenges that can only be done in a clinical setting and their application in nutrition studies therefore seems less feasible.

Fig. 4 The time course of plasma cytokine concentrations after an inflammatory challenge (in this case a bolus intravenous injection of Escherichia coli lipopolysaccharide (LPS)). Concentrations and time are shown on arbitrary scales. There is an initial appearance of inflammatory cytokines (TNF and IL-1), followed by IL-6, and then anti-inflammatory cytokines (TNF receptor (TNFR), IL-1 receptor antagonist (IL-1ra) and IL-10). Reproduced from Bentham Science Publishers(Reference Andreasen, Krabbe and Krogh-Madsen300) with permission.
Early response to (the adjuvant component of) vaccines
Effects of vaccination are primarily studied in relation to enhancing vaccination efficacy and are evaluated according to the level of protective antibody levels. However, innate immune factors including pro-inflammatory cytokines are induced during the (early) response to vaccination. These early responses are probably induced by the adjuvants used rather than by the antigen that is provided in the vaccine. Even though early responses to vaccination have not been studied extensively, several studies have indicated that enhanced levels of pro-inflammatory cytokines can be detected in serum post-vaccination. These studies have, however, been performed in different study populations, with different vaccines using different adjuvants (although the seasonal influenza vaccine has been used frequently), and serum samples have been evaluated at different time points after vaccination, ranging from 2 h to several weeks. Even so, in six of seven studies, IL-6 was increased after vaccination. This occurred 2–3 h after vaccination(Reference Harrison, Brydon and Walker199), but could also be detected after 1 or 2 d(Reference Carty, Heagerty and Nakayama200, Reference Lanza, Barone and Scalone201) or even after several weeks(Reference Trzonkowski, Mysliwska and Pawelec202). In contrast, no change in IL-6 2 weeks after vaccination was noted by Glaser et al. (Reference Glaser, Robles and Sheridan203). TNF was not increased 3 h after vaccination in a single study(Reference Wright, Strike and Brydon204), and CRP was increased in three vaccination studies(Reference Carty, Heagerty and Nakayama200, Reference Lanza, Barone and Scalone201, Reference Werba, Veglia and Amato205). Other markers that have been described to increase after vaccination are MCP-1, eotaxin and IL-13(Reference Dehqanzada, Storrer and Hueman206), as well as IL-2, IL-12 and IL-15(Reference Mysliwska, Trzonkowski and Szmit207). As noted for other challenge models, in some studies, it was clear that individuals with pre-existing conditions (inflammatory, stress, depression or age-related IL-6 increase) had more prominent pro-inflammatory cytokine responses than the controls in the same studies(Reference Harrison, Brydon and Walker199–Reference Mysliwska, Trzonkowski and Szmit207). These findings have indicated that pro-inflammatory cytokine levels can be measured after vaccination; however, more studies are needed to standardise a vaccination approach as an inflammatory challenge. Feasibility is expected to be high, and the responses noted seem quite reproducible, suggesting that most persons tested will indeed respond by the production of pro-inflammatory mediators. Using vaccination as an inflammatory challenge could be relevant to identify anti-inflammatory effects of nutritional intervention. An added advantage is that protective antibody titres can be used as a readout at a later stage so that a single study can be used to measure two different immunological outcomes (inflammation and immune response). However, it should be noted that pre-existing elevated baseline levels of pro-inflammatory cytokines are associated with decreased vaccination responses(Reference Trzonkowski, Mysliwska and Szmit208).
Skin tests
Delayed-type hypersensitivity and contact hypersensitivity responses to locally applied antigens or contact allergens can be used to assess local inflammatory responses triggered by the specific response to the antigen or allergen. These responses rarely result in measurable effects on circulating cytokines or acute-phase proteins, but instead can be measured as local erythema or epidermal induration 24–48 h after application. As the skin is easily accessible, local inflammation can be studied in great detail using biopsies. Delayed-type hypersensitivity and contact hypersensitivity responses can only be elicited in sensitised subjects and rely on the uncontrolled history of exposure resulting in large variation among individuals and in many non-responders. This is key to their diagnostic value, but hampers application as a challenge model in nutrition studies. Nonetheless, in the past, studies have successfully assessed the impact of nutrition on delayed-type hypersensitivity responses using the cell-mediated immunity multitest to simultaneously administer multiple antigens(Reference Meydani, Barklund and Liu209–Reference Wolvers, van Herpen-Broekmans and Logman211). Unfortunately, this device is no longer available and no robust alternative methods have been described for routine application of multiple antigens. Using a single Candida antigen, delayed-type hypersensitivity responses have recently been shown to be associated with a systemic inflammatory state in older subjects (aged 60–85 years), with positive responders having significantly lower serum CRP levels(Reference Pence, Lowder and Keylock212). To control both the sensitisation and elicitation phase, studies have also explored the use of experimental sensitisation to an uncommon contact sensitiser, diphenylcyclopropenone, that would not normally be encountered(Reference Zachariae, Jorgensen and Christensen213, Reference Sleijffers, Garssen and de Gruijl214). Although there are ethical considerations, this has been successfully used to assess the effect of (exercise) intervention on both the sensitisation and elicitation phase using induration due to the local inflammation of the skin as the readout(Reference Harper Smith, Coakley and Ward215).
Tissue damage
Exercise challenges
It is well established that strenuous exercise (either an acute intensive bout or longer duration such as marathon running) increases the concentrations of leucocytes and neutrophils immediately following the exercise and induces an increase in inflammatory marker concentrations in the bloodstream(Reference Pedersen and Hoffman-Goetz216). However, if a challenge model should be applicable to the general population to assess the impact of (dietary) interventions on the elicited inflammatory response, then a moderate exercise challenge may be more relevant. The effects of moderate aerobic exercise on pro-inflammatory cytokine production are less well characterised than the effects of strenuous exercise as discussed earlier. Most data from exercise studies relate to long-term aerobic training (in which there is an anti-inflammatory effect) and to strenuous exercise such as marathon running (in which pro-inflammatory cytokines, especially IL-6, are elevated), as discussed elsewhere(Reference Walsh, Gleeson and Shephard87, Reference Walsh, Gleeson and Pyne88). Increases in such inflammatory cytokines after short-term exercise are much less pronounced. The production of IL-6 increased after mild exercise in studies performed in healthy as well as diseased adults(Reference Castellano, Patel and White217–Reference van Helvoort, van de Pol and Heijdra221), but was unchanged in other studies(Reference Mercken, Gosker and Rutten222, Reference Rabinovich, Figueras and Ardite223). Whereas muscles produce the pro-inflammatory IL-6 during acute bouts of exercise; paradoxically, IL-6 also has anti-inflammatory effects by promoting the release of IL-1 receptor antagonist and IL-10(Reference Walsh, Gleeson and Shephard87). This may in part explain the notion that regular, mild, low-impact exercise has anti-inflammatory effects. The production of TNF remained unchanged after mild exercise in healthy as well as diseased adults in three studies(Reference Lucia, Smith and Naidoo220, Reference Mercken, Gosker and Rutten222, Reference Ionescu, Mickleborough and Bolton224) and increased in another study(Reference Kinugawa, Kato and Ogino218). The numbers of circulating total leucocytes, lymphocytes and monocytes increased after mild exercise(Reference van Helvoort, Heijdra and de Boer225). Interestingly, in some studies, inflammatory cytokines increased after moderate exercise in patients, but not in control subjects. Rabinovich et al. (Reference Rabinovich, Figueras and Ardite223) described that serum TNF increased in patients with cystic fibrosis after moderate exercise but not in controls. Likewise, IL-6 increased after mild exercise in cystic fibrosis patients, but not in healthy controls in another study(Reference Ionescu, Mickleborough and Bolton224). The effect of mild exercise on CRP was only tested in a single study, and was found to be increased in patients with coronary artery disease but not in control subjects(Reference Kop, Weissman and Zhu219). Similar findings have been seen in studies with children(Reference Ploeger, Takken and de Greef226).
UV irradiation
Whole-body exposure to high doses of sunlight or UVB radiation causing sunburn increases the levels of inflammatory mediators in the circulation. Milder or more focused exposure to UVB radiation was shown to decrease the skin contact hypersensitivity response(Reference Sleijffers, Garssen and de Gruijl214) as well as some systemic responses such as natural killer cell activity(Reference Sleijffers, Garssen and de Gruijl214, Reference Gilmour, Vestey and George227) and ex vivo production of cytokines(Reference Jones, Guckian and el-Ghorr228). However, circulating cytokine levels after single exposure to UVB were not affected(Reference McLoone, Man and Yule229, Reference Narbutt, Lesiak and Skibinska230) and even after 10 d of repeated exposure, only IL-8 and TNF were marginally increased(Reference Narbutt, Lesiak and Skibinska230). It is therefore unlikely that a practically feasible UV challenge can be developed that can be used to assess indicators of inflammation systemically. However, using microdialysis and/or punch biopsies, pro- and anti-inflammatory responses can be sequentially assessed in the skin in the hours to days following a single dose of two or three minimal erythemal doses of UVB radiation(Reference Angst, Clark and Carvalho231–Reference Brink, Szamel and Young233). This is a useful model to assess local mediators including short-lived eicosanoids involved in the initiation and resolution phase of UVB-induced skin inflammation(Reference Rhodes, Gledhill and Masoodi234).
General comments on challenge models
CRP, TNF and IL-6 are the most commonly measured inflammatory markers after the various challenges, although other cytokines, chemokines, adhesion molecules and leucocyte numbers have also been used. For challenges that induce inflammation in the skin, local markers such as swelling, erythema, cellular infiltrate and local levels of mediators in exudate from (suction) blisters or biopsies have also been used. Steady-state differences in inflammatory markers between patients with diseases with an inflammatory component and healthy people seem to be more consistent than differences in post-challenge responses. However, stronger or more extended inflammatory responses (IL-6, TNF and CRP) to challenges in patients v. controls have also been noted in several studies. Challenges other than with glucose are not, or are poorly, standardised and differ greatly between studies with respect to the challenge (amount and quality of fats, type and duration of exercise, wavelength and energy intensity of UV radiation, and type of vaccine), markers measured, time points measured and the way in which results are analysed. In most studies, the inflammatory response is assessed at one or more arbitrary time points after the triggering challenge, few studies have followed the detailed kinetics of the responses, few included a proper time control for circadian rhythms and very few have integrated the entire response, for instance, by analysing the AUC(Reference Erikstrup, Ullum and Pedersen197). Different challenges have been used in different contexts, using different sets of readouts and different (subject or patient) populations. This makes it complex to compare the challenges based on available data. A recent study observed that multiple intermediates of the glutathione synthesis pathway showed time-dependent suppression in response to the OGTT in overweight subjects(Reference Wopereis, Rubingh and van Erk235). These metabolic perturbations were subtle and were only observed using metabolic profiling at multiple time points following a physiological challenge (OGTT). This is a good example of more sophisticated integrative analysis, which may also be very useful for the detailed analysis of the kinetics of complex inflammatory responses(Reference Wopereis, Rubingh and van Erk235).
It would be very useful to have reliable kinetic data of the inflammatory responses following well-selected, standardised pro-inflammatory challenges. These could then serve as a starting point for systematic comparisons of the kinetic profiles in different subpopulations (obese, insulin insensitive, diabetic, older individuals, etc.) and for careful evaluations of the impact of (dietary) interventions on the resilience of these responses. The established kinetic responses following an intravenous LPS challenge could serve as a useful point of reference for this (see Fig. 4). However, care should be taken not to introduce artifacts due to the sampling as several studies have demonstrated that the mere presence of an indwelling catheter results in the continued local elevation of IL-6 levels compared with samples taken from the contralateral arm using repeated venepuncture(Reference Seiler, Muller and Hiemke236–Reference Haack, Reichenberg and Kraus238).
Although it may not yet be possible with the relatively limited dataset available, it would be very useful to work towards standardised challenges combined with standardised sampling and analytical regimens and standardised data analyses, and use these as an integrated biomarker of inflammatory resilience. This resembles what has been done with the OGTT for the assessment of insulin resistance and diabetes and would facilitate comparison between studies in prospective cohorts, which is needed to establish the predictive value and clinical relevance of resilience to (acute) inflammatory challenges.
Conceptually, the use of standardised challenges is an interesting approach and there are some indications that resilience of dynamic inflammatory responses may have added value over assessing markers during steady state only. However, there are too few data to draw firm conclusions at this point. Some studies have indicated different post-challenge inflammatory responses in patient populations, but it is not yet feasible to define healthy inflammatory responsiveness to a particular challenge based on the available published data. Oral glucose and fat challenges and potentially the early response to a vaccine seem to be best standardised, most relevant and most feasible challenges for application in nutrition studies and for inclusion in prospective cohort studies.
Key messages
(1) Inflammatory responsiveness or resilience to challenges may provide a more sensitive and meaningful indication of inflammatory state in the general population than the assessment of markers during steady state.
(2) There is a need to standardise challenge(s) and assessment of the subsequent inflammatory response, including multiple sampling points to assess the kinetics of the response. As many inflammatory markers also have a circadian rhythm, such studies should involve proper time controls.
(3) There is a need to firmly establish whether standardised assessment of the dynamic response to an (acute) inflammatory challenge does indeed add value (clinical relevance, sensitivity, relevance, interpretability, etc.) compared with assessment of inflammatory markers during the steady state.
(4) It is likely that a combination of multiple inflammatory markers (perhaps the same as those used to assess inflammation in the steady state) and integrated readouts based upon kinetic analysis following defined inflammatory challenges will be most informative.
Emerging markers of inflammation
Expression of cell surface markers
New technologies allow the profiling of hundreds of cell surface markers in peripheral blood and tissues, and the identification of surface markers whose extent of expression correlates with the inflammatory status. Recent studies have suggested that surface expression of CD11b on granulocytes is a marker of granulocyte activation; its expression is increased in obesity(Reference Viardot, Lord and Samaras239). Weight loss in obese subjects and in individuals with diabetes is associated with decreased expression of CD11b on granulocytes and of CD66b on monocytes(Reference Viardot, Lord and Samaras239). CD11c, a β2 integrin expressed on monocytes, plays an important role in monocyte adhesion to inflamed endothelial cells. Obese human subjects with the metabolic syndrome have higher CD11c levels on monocytes than lean healthy controls(Reference Wu, Perrard and Wang240), suggesting that these monocytes are involved in the inflammatory process of atherosclerosis. For these biomarkers, little information is available on the impact of confounders, such as smoking or physical (in)activity.
Clusters of markers
An important approach to ascertaining the clinical relevance of selected biomarkers of inflammation is to perform longitudinal studies following human subjects over time(Reference Derhovanessian, Larbi and Pawelec241). Several such longitudinal studies in human subjects have sought to identify biomarkers of morbidity and mortality, with mortality as the most ‘robust clinical endpoint’. Inflammatory factors such as CRP, TNF and IL-6 have repeatedly been found to associate with increased mortality and frailty, but mostly in cross-sectional studies(Reference Pawelec, Derhovanessian and Larbi242, Reference Pawelec, Akbar and Caruso243). Moreover, analyses of associations with single factors in either cross-sectional or longitudinal studies are usually weak or not always reproduced in different populations. A major challenge in studies of long-lived species like humans is clearly logistics of long-term follow-up. An approach to this was made in the Swedish immune longitudinal studies including octo- and nonagenarians (OCTO/NONA) studies, taken here as an example, which examined soluble and cellular markers at baseline in 85-year-old subjects to seek predictive biomarkers for mortality in cohorts with high mortality over reasonable follow-up times(Reference Wikby, Ferguson and Forsey244, Reference Wikby, Nilsson and Forsey245). The rationale was to identify those predictive biomarkers with potential applicability to other, younger, populations. These studies not only performed follow-up at 2, 4 and 6 years, but also resampled, so that changes to biomarkers over time could be assessed. It was found that single markers were relatively uninformative, as expected from the above considerations, but that clusters of markers were significantly associated with mortality. Thus, a cluster of cellular markers including an inverted CD4:CD8 ratio due to an accumulation of CD8+ late-stage memory cells and infection with the common herpes virus cytomegalovirus, now thought to drive this ‘memory inflation’, were significantly associated with mortality. Cytomegalovirus infection has a pro-inflammatory action, increasing the levels of cytokines such as interferon-γ. The OCTO/NONA studies also identified a second cluster of soluble factors comprising increased IL-6, CRP and the thyroid hormone and vitamin A transport protein transthyretin, which correlated with cognitive impairment and independently associated with mortality. Individuals with neither risk cluster had better survival at follow-up than those with either one or the other risk cluster, and people simultaneously with both risk clusters had the worst survival(Reference Wikby, Ferguson and Forsey244). These findings emphasise that multiple inflammatory factors need to be assessed in combination. The importance of the balance between pro- and anti-inflammatory factors was emphasised by the finding that although the majority of subjects in this risk group were already deceased at 2- and 4-year follow-up, a minority exited the risk group and remained alive at this time. These rare individuals were characterised by high plasma IL-10 levels, as well as retaining high IL-6 levels(Reference Wikby, Nilsson and Forsey245). This risk cluster remains an emerging marker because, as defined thus far, it has limitations that require further study applying more sophisticated methodology to well-characterised cohorts. These limitations include, but are not limited to, the following: the cluster has only been shown to be relevant in a small study in one very old population in one country; it lacks the sophistication of modern immune analysis; it is unknown at which calendar age it becomes relevant; associations with cause of death are unknown and associations with underlying disease states are also unknown; finally, it does not take nutrition, psychological stress or pathogen environment into account. Given that nutrients and dietary patterns may influence the components of the clusters described above(Reference Calder, Albers and Antoine1, Reference Calder, Ahluwalia and Brouns6), it is likely that the clusters themselves may be targets for nutritional interventions.
Stress markers
The adverse tissue environment of immune-mediated and metabolic disease triggers a cellular stress programme in response to hypoxia, inflammation, reactive oxygen species as well as the lack or imbalanced presence of nutrients(Reference Rath and Haller246). Specific metabolic and functional processes occur in specialised cellular organelles such as the ER, peroxisomes and mitochondria(Reference Yoshida247). The unfolded protein response (UPR) is a well-conserved mechanism of these organelles to restore cellular homeostasis in response to stress; however, unresolved or chronic activation of UPR pathways induces inflammatory mechanisms or cell death. Even though they are phenotypically different, the diseases described in Table 4 share similarities of the cell stress response at the molecular level(Reference Renz, von Mitius and Brandtzaeg248). Hypoxia and inflammatory signals as well as metabolic challenges increase cellular energy requirements and reactive oxygen species production and have a negative impact on protein folding machinery of the cell. Consistently, interference with ER(Reference Bertolotti, Wang and Novoa249–Reference Kaser, Lee and Franke251) and mitochondrial UPR-associated signalling pathways(Reference Haga, Saito and Tsukumo252–Reference Rath, Berger and Messlik254) has been shown to have an impact on disease susceptibility and pathological outcome in various animal models. Generally accepted surrogate markers for UPR include cellular expression and secretion of compartment-specific chaperones (CPN) such as glucose-related protein (GRP) 78 and heat shock protein (HSP) 78 CPN60, the latter representing the main CPN involved in protein folding in the mitochondria, since induction of these proteins is a main purpose of UPR signalling(Reference Schroder and Kaufman255, Reference Zhao, Wang and Levichkin256). Increased protein and mRNA expression levels of GRP78 in human mononuclear cells were shown to be associated with deregulated blood glucose homeostasis, increased levels of inflammatory cytokines and the metabolic syndrome, demonstrating clinical relevance for ER-UPR markers in human disease(Reference Sage, Holtby-Ottenhof and Shi257). In addition, plasma HSP60 levels were associated with hypertension and increased risk for CVD(Reference Pockley, Wu and Lemne258, Reference Pockley, De Faire and Kiessling259). To evaluate ER-derived signalling leading to altered gene expression, splicing of X-box binding protein 1 (Xbp-1) mRNA is commonly monitored. The spliced mRNA encodes an active, UPR-associated transcription factor, spliced X-box binding protein 1 (sXBP-1), and splicing of Xbp-1 mRNA represents an early event during ER-UPR(Reference Kaser, Lee and Franke251, Reference Schroder and Kaufman255). Induction of GRP78 or CPN60 is mostly measured by Western blot analysis, immunohistochemical analysis and quantitative polymerase chain reaction (PCR), whereas Xbp-1 mRNA splicing is detected by using specific primers for Xbp-1 mRNA or sXbp-1 mRNA for quantitative PCR analysis. In conclusion, GRP78 and HSP60 represent surrogate markers for chronic conditions with inflammatory pathologies that can be measured in biopsies from the disease-relevant tissue or in specifically isolated target cells as well as circulating protein in plasma(Reference Renz, von Mitius and Brandtzaeg248). These could be tested as potential early markers for the onset of inflammation, where data are not yet available, including evaluating the potential for modulation by interventions.
MicroRNA
MicroRNA (miRNA) are short (average twenty-two nucleotides) non-coding ribonucleic acids that have been found in eukaryotic cells. They have important regulatory functions by controlling gene expression, by inhibiting protein translation and by inducing target mRNA destabilisation(Reference Bartel260). miRNA can be detected by PCR, specific microarrays or, more comprehensively, by next-generation sequencing approaches(Reference Buermans, Ariyurek and van Ommen261). Endogenous circulating miRNA in human plasma are potential candidates as stable biomarkers for health status, organ dysfunction and disease(Reference Fichtlscherer, Zeiher and Dimmeler262–Reference Vickers, Palmisano and Shoucri264). As many of the common diseases have an inflammatory component that may be regulated by non-coding RNA(Reference O'Connell, Rao and Baltimore265), it is expected that plasma miRNA profiling will reveal sensitive biomarkers for the onset or modulation of chronic inflammation in humans(Reference Fichtlscherer, Zeiher and Dimmeler262, Reference Zahm, Thayu and Hand266–Reference Fichtlscherer, De Rosa and Fox268). The latter could include nutritional modulation.
Microbiota and associated markers
As discussed earlier, microbiota and, particularly, gut microbiota might play an important role in human health and physiopathology. It is possible that circulating traces of translocated bacteria or bacterial components could be used as biomarkers to predict metabolic disease risk in humans. Measurements of blood LPS have frequently been reported in the literature as a feature of mucosal barrier failure. However, such measures are rather unspecific, are limited to Gram-negative organisms and pose some technical difficulties. Alternative methods to detect material of bacterial origin or to detect responses to specific organisms are emerging. Indeed, a battery of antibodies against different bacterial antigens is currently under investigation, although their physiological role is still unclear. However, this approach may provide new markers of inflammation. For instance, an array of immunohistological tools have been developed to study, diagnose and/or score ulcerative colitis and Crohn's disease(Reference Bossuyt269, Reference Dotan270). So far, the combination of perinuclear anti-neutrophil cytoplasmic antibodies (P-ANCA) and anti-Saccharomyces cerevisiae antibodies (ASCA) seems to be the best serological test to distinguish ulcerative colitis (P-ANCA+ and ASCA− /low) from Crohn's disease (P-ANCA− /low and ASCA+)(Reference Bossuyt269). The usefulness of such tools in the generally healthy population needs further development. In addition, specific bacterial 16S ribosomal DNA detection by PCR amplification in blood is a tool being used more frequently(Reference Reesink, Mohammadi and Pietersz271–Reference Rajendhran and Gunasekaran273). The combination of excellent sensitivity and specificity, ease and speed has made real-time PCR technology an appealing alternative to conventional culture-based testing methods. This approach has been used recently to show a positive association between 16S ribosomal DNA in blood and diabetes onset(Reference Amar, Serino and Lange274). The 16S ribosomal DNA concentration was analysed in blood in a cohort of 3280 subjects without diabetes or obesity. Baseline 16S ribosomal DNA level was higher in those subjects who developed diabetes by the 9-year follow-up(Reference Amar, Serino and Lange274).
Key messages
(1) Altered expression of activation markers on the surface of circulating leucocytes may be biomarkers of inflammation.
(2) Longitudinal studies of the very old suggest that a profile of plasma inflammatory markers correlates with altered risk of mortality and is more predictive than any marker alone.
(3) UPR are the consequence of sustained cellular stress in the context of immune-mediated or metabolically driven chronic pathologies with GRP78 and CPN60 as surrogate markers.
(4) Endogenous circulating miRNA are quite stable potential biomarkers for health status and organ dysfunctions, and may reveal very sensitive information about inflammatory state.
(5) There are emerging approaches to assess the microbiota and the translocation of micro-organisms.
(6) These emerging markers of inflammation may be influenced by nutrition and represent potential novel targets for nutritional interventions.
Emerging technologies
Applications of omic technologies to identify new markers and to monitor the effect of interventions
Genomics was originally described as the comprehensive analysis of the genome of an organism that includes efforts to determine the entire DNA sequence and fine-scale genetic mapping. A broader definition than this may also be used(275), according to which ‘genomics’ is the study of all of the genes of a cell, or tissue, at the DNA (epigenome, genome), mRNA (transcriptome) and protein (proteome) levels. New high-throughput technologies such transcriptomics, proteomics and metabolomics have created unprecedented opportunities for a less biased and more comprehensive assessment of even subtle phenotypic changes. To integrate applications of these technologies more efficiently into nutrition research, nutrigenomics has been initiated as a new innovative research strategy about 10 years ago when the working draft of the human genome was announced(Reference Afman and Müller276–Reference Afman and Müller280). In addition to comprehensive phenotyping, nutrigenomics includes innovative combinations of molecular nutrition research and genomics applications. It allows the comprehensive study of diverse tissue and organ-specific gene expression patterns (transcriptome), organisation and modification of the chromatin structure (epigenome), protein expression patterns including post-translational modifications (proteome) as well as metabolite profiles (metabolome)(Reference Wittwer, Rubio-Aliaga and Hoeft279, Reference Kussmann and Van Bladeren281). To achieve the powerful integration of all different nutrigenomic and phenotype data systems, biology strategies are being developed. Nutritional systems biology is a holistic approach merging various omics data (e.g. genome, epigenome, metabolome, proteome, microbiome) from individual organs and body compartments at different time points of a nutritional intervention with the use of computational methods and predictive mathematical models. This requires a large degree of standardisation, multidisciplinary collaboration, improved bioinformatics and statistical methods and sophisticated and partly Web-based databases such as the Nutritional Phenotype database(Reference Bouwman, Vogels and Wopereis282, Reference van Ommen, Bouwman and Dragsted283).
It is becoming evident from recent studies that nutrigenomics is promoting an increased understanding of how a nutritional intervention mechanistically influences metabolic pathways and homeostatic control. Furthermore, it reveals how this regulation is disturbed during development of diseases, and the extent to which individual sensitising genotypes contribute to such diseases by allowing comprehensive phenotyping and the identification of early biomarkers for pre-disease states. A few recent nutrigenomics-based applications related to systemic and organ-related pro-inflammatory states are discussed here. Nutrigenomics studies using appropriate mouse models are essential to better understand the systems-wide changes of a metabolically healthy to a less healthy organism and to identify potential early biomarkers that can then be validated in human studies(Reference de Wit, Boekschoten and Bachmair284–Reference Duval, Thissen and Keshtkar286). Recent studies have already shown the great opportunities of this new research area(Reference Frood191, Reference Grayson287, Reference Laursen288).
Whole genome transcriptome analysis has been used to demonstrate that intake of n-3 fatty acids by healthy older persons for 26 weeks can alter the gene expression profiles of peripheral blood mononuclear cells, as a ‘surrogate’ biomarker for whole-body health status, to a more anti-inflammatory and anti-atherogenic status, although CRP was not changed(Reference Bouwens, van de Rest and Dellschaft289). Pathway analyses revealed significantly decreased expression of genes involved in inflammatory pathways, such as eicosanoid synthesis, cytokine signalling and mitogen-activated protein kinase signalling, and in pathways related to atherosclerotic processes, such as cell adhesion and scavenger receptor activity(Reference Bouwens, van de Rest and Dellschaft289). A combination of peripheral blood mononuclear cells and adipose tissue transcriptome, plasma protein analysis and plasma and urine metabolomics was used to study the effects of specific dietary components to reduce low-grade inflammation as well as metabolic and oxidative stress in healthy overweight men with mildly elevated plasma CRP levels(Reference Bakker, van Erk and Pellis290). Whereas CRP levels were again found to be unchanged, a multitude of subtle changes were detected by an integrated analysis of nutrigenomics datasets, which indicated modulated inflammation of adipose tissue, improved endothelial function, altered oxidative stress and increased liver fatty acid oxidation. Another nutrigenomics study investigated the effect of saturated and monounsaturated fat-rich diets on insulin sensitivity, serum lipids and gene expression profiles of adipose tissue in subjects at risk of the metabolic syndrome(Reference van Dijk, Feskens and Bos291). Consumption of the saturated fat-rich diet resulted in the up-regulation of processes related to inflammation. Several genes linked to inflammation included markers for recruitment of leucocytes, genes involved in eicosanoid synthesis and monocyte/macrophage markers. The monounsaturated fat-rich diet led to a more anti-inflammatory gene expression profile and resulted in a down-regulation of several adipose tissue macrophage genes. This study showed that subtle organ-specific changes in phenotypic profiles could be detected with genomics methods, which is very relevant to discriminate between early organ-related changes and late systemic changes in (pro-)inflammatory and less healthy status.
In a more recent study, a large set of plasma proteins related to chronic diseases, inflammation, endothelial function and metabolic signalling was assessed in a population of relatively healthy, middle-aged, overweight subjects(Reference van Dijk, Feskens and Bos291, Reference van Dijk, Feskens and Heidema292). Clusters of proteins associated with BMI or insulin were identified. Leptin and a number of pro-inflammatory proteins, previously identified as possible biomarkers for obesity-related disease (e.g. C3, CRP, serum amyloid P, vascular endothelial growth factor), clustered together and were positively associated with BMI and insulin. IL-3 and IL-13 clustered together with apoA1 and were inversely associated with BMI, and might be potential new biomarkers. These plasma protein clusters could have potential applications for improved phenotypic characterisation of subjects in nutritional intervention studies or as biomarkers in the early detection of obesity-linked disease development and progression.
As the recent developments in using nutrigenomics for metabolic profiling and characterising complex interactions are looking very promising, future studies have to link these results to accepted functional outcome measures. Application of metabolic perturbations in combination with comprehensive nutrigenomics-based phenotyping will improve the diagnostic sensitivity and ultimately allow discriminating between healthy, less healthy and unhealthy people(Reference van Ommen, Keijer and Heil181, Reference Wopereis, Rubingh and van Erk235, Reference van Ommen, Keijer and Kleemann293).
Non-invasive imaging
Recent developments in non-invasive molecular imaging (such as magnetic resonance imaging) are allowing quantification of physiological changes in vivo using specific probes. Nanoprobes can be used to image specific cells and tissues within a whole organism, at the moment mainly in animal models such as mice(Reference Minchin and Martin294). Intravital fluorescence microscopy in mice is beginning to provide quantitative and dynamic insights into cell biology and immunology(Reference Pittet and Weissleder295). Recent application of specific mass spectrometry techniques in combination with magnetic resonance imaging has provided a systems biology approach to study inflammation in mice(Reference Attia, Schroeder and Seeley296). In a recent study, novel microscopy techniques have been used to characterise the composition of visceral adipose tissues of lean and obese mice including adipocytes, macrophages and collagen fibrils in a label-free manner, allowing dynamical monitoring of lipid metabolism-related processes relevant for immunometabolism(Reference Urasaki, Johlfs and Fiscus297). Finally, non-invasive imaging has been applied in mouse models of inflammatory disease and for imaging of macrophages in vivo by using activatable fluorescent probes, radiolabelled inhibitors and nanoparticles(Reference Quillard, Croce and Jaffer298). Increasingly, these imaging techniques are also discussed for their potential to identify subclinical atherosclerosis in humans(Reference Fuster, Lois and Franco299). The goals for the years to come must include translation of the experimental work to visualisation of biological targets relevant for local organ-related and systemic inflammatory states in humans.
Key messages
(1) Applications of metabolic perturbations in combination with comprehensive nutrigenomics-based phenotyping will significantly improve the diagnostic sensitivity, allowing discrimination between healthy, less healthy and unhealthy people.
(2) For predictive mathematical modelling of the modulation of inflammatory states by nutritional interventions, the integration of different nutrigenomics and phenotype data from individual organs and body compartments has to be achieved by nutritional systems biology approaches.
(3) A range of imaging techniques is emerging that have the potential to non-invasively identify inflammation and its consequences.
Summary, conclusions, identification of key gaps, overall recommendations and overall key messages
There is an urgent necessity for reliably monitoring inflammatory status in the context of disease and interventions to prevent or treat disease, including dietary modification. Currently, guidelines on how to accurately assess inflammation are lacking. To monitor inflammation in a meaningful way, the markers used must be valid; that is, they must reflect the inflammatory process under study and they must be predictive of future health status. Inflammation per se is a normal, essential, complex physiological process crucial for restoring and maintaining health. This process involves many cells and mediators. The latter include numerous peptides and proteins that circulate in the bloodstream (e.g. cytokines, chemokines, soluble forms of adhesion molecules and acute-phase proteins). These are involved in, or produced as a result of, the inflammatory process irrespective of its trigger and its location. Thus, they are common to all inflammatory diseases, to acute and chronic inflammatory responses and to both high-grade and low-grade inflammation. They would be considered as general markers of inflammation, with no specificity with regard to the trigger or the site of inflammatory activity. Furthermore, many of these soluble markers are present at readily detectable concentrations in the bloodstream of healthy individuals, although it is not always clear what their role in healthy persons might be. In contrast, there are inflammatory markers specific to inflammatory disease, which cannot usually be measured, or are present at very low concentrations, in healthy individuals. The focus in inflammation research has been very much on such pro-inflammatory markers that are part of an amplification cascade or that cause tissue damage, and the factors that influence these and which can be used to distinguish disease. There has been less emphasis on anti-inflammatory markers, which include receptor antagonists, soluble forms of receptors, cytokines and lipid mediators that act to down-regulate the production of inflammatory mediators. There is even less understanding of those factors that terminate (resolve) inflammation. Currently, there is no consensus on which markers of inflammation best represent low-grade inflammation or can differentiate between acute and chronic inflammation or among the initiation, propagation and resolution phases of inflammatory responses. For most inflammatory markers, there is limited information on the predictive value of (differences in or changes in) their concentration from prospective studies; that is, information on their role as (independent) markers of disease risk and outcome is largely lacking. Many modifying factors affect the concentration of an inflammatory marker at a given time; the effects of some factors, such as age, physical (in)activity and smoking, on a small number of inflammatory markers are well described, but the effects of these factors on the range of markers considered here are mostly not known. In particular, knowledge of the effects of genotype, sex and gut microbiota is limited. In addition, those modifying factors for which there are relatively few data (e.g. genotype) may have a larger effect on inflammatory responses than factors for which there are larger amounts of data available. Measuring the concentration of inflammatory markers in the bloodstream under basal conditions (e.g. in fasted blood in the early morning) is probably not as informative as data related to the concentration change in response to a challenge. In the literature, a number of challenges reflecting metabolic stress, infection and tissue damage have been described. However, many of these challenges are poorly standardised, and the nature of the dynamic of the response, the markers influenced (or not), the extent of variation in the response among individuals and the effect of modifiers on the response are currently not well documented. Efforts to standardise pro-inflammatory challenge tests to assess the resilience of inflammatory control are urgently needed. Most intervention studies focus on a single marker, or on a small number of markers, of inflammation. There are few attempts to define composite markers that may be more specific or predictive. Patterns and clusters may be more robust biomarkers of inflammatory state and inflammatory response than individual or small numbers of markers. Therefore, it is likely that a combination of multiple inflammatory markers (perhaps the same as those used to assess inflammation in the steady state) and integrated readouts based on kinetic analysis following defined challenges will be the most informative biomarker of inflammation. New techniques are emerging to monitor inflammation but these are, in general, at the early experimental stages and have had little translation to humans. There will be a need to fully validate these technologies.
Overall key messages
(1) Acute inflammation is a normal physiological response crucial for maintaining homeostatic control, but when it becomes chronic, inflammation contributes to the pathophysiology of a range of diseases.
(2) Common soluble markers of inflammation include cytokines, chemokines, lipids, acute-phase proteins and adhesion molecules and common blood cellular markers of inflammation include various subclasses of leucocytes.
(3) The same markers are involved in both acute and chronic inflammatory processes, meaning that the markers themselves, in isolation, cannot be used to distinguish the two processes.
(4) There are a number of endogenous and exogenous modifiers that influence the levels of inflammatory markers resulting in significant between-individual variation. These modifiers include age, body fatness, physical (in)activity, sex, genotype, smoking habits, diet, the composition of the gut microbiota and the use of certain medications.
(5) Inflammatory responsiveness or resilience to challenges may provide a more sensitive and meaningful indication of inflammatory state in the general population than the assessment of markers during the steady state.
(6) There is a need to standardise pro-inflammatory challenge(s) and assessment of the subsequent inflammatory response.
(7) It is likely that a combination of multiple inflammatory markers (perhaps the same as those used to assess inflammation in the steady state) and integrated readouts based on kinetic analysis following defined challenges will be the most informative biomarker of inflammation.
(8) A number of novel markers of inflammation are emerging but these require further exploration and validation.
Acknowledgements
This work was commissioned by the Nutrition and Immunity Task Force of the European branch of the International Life Sciences Institute (ILSI Europe). Industry members of this task force are Danone, Institut Mérieux, Nestlé, Pfizer Consumer Healthcare, FrieslandCampina, Seven Seas, Südzucker/BENEO Group, Unilever and Yakult Europe. This publication was coordinated by Lena Jönnson and Marie Latulippe, Scientific Project Managers at ILSI Europe. For further information about ILSI Europe, please email info@ilsieurope.be or call +32 2 771 00 14. The opinions expressed herein and the conclusions of this publication are those of the authors and do not necessarily represent the views of ILSI Europe nor those of its member companies. A draft of this report was discussed at a workshop held in Seville, Spain from 14 to 16 May 2012. In addition to the authors, the following individuals attended the workshop and contributed to the discussions: Jalil Benyacoub, Nestlé Research Center, Switzerland; Deborah Braun, Institut Mérieux, France; Maria Denis, Biomedical Sciences Research Centre; Alexander Fleming, Greece; Kevin Fritsche, University of Missouri, USA; Ellen Kreijveld, Unilever, The Netherlands; Jose Lopez Miranda, University of Cordoba, Spain; Anna Nicolaou, University of Bradford, UK; Fátima Pérez de Heredia, Spanish National Research Council, Spain; André Siemensma, FrieslandCampina, The Netherlands; Cathy Signoret, Cosucra Groupe Warcoing, Belgium; Ben van Ommen, TNO, The Netherlands; Willem Vas Dias, Seven Seas, UK; Stéphane Vidry, ILSI Europe, Belgium; Neil Walsh, Bangor University, UK; Danielle Wolvers, Unilever, The Netherlands. The authors thank these individuals for their valuable input. P. C. C. has research funding from Beneo-Orafti, Beghin-Meiji, Abbott Nutrition and ViforPharma and is a consultant to Danone. R. A. was an employee of Unilever R&D until 2012 and has been an employee of NutriLeads since that time. N. B. is an employee of Nestlé Research Center. R. B.-S. is an employee of Danone. L. S. J. and M. E. L. are employees of ILSI Europe. J. M. is an employee of Pfizer. C. M. is an employee of Institut Mérieux. M. M. and A. M. are government employees and have no conflicts of interest to declare. G. P. was a consultant to Unilever Central Research. R. J. J. v. N. is an employee of FrieslandCampina. J. Z. is an employee of Yakult Europe. The remaining authors have no conflicts of interest. All authors contributed to discussions and had input into writing the article. All authors read and approved the final version of the article.











