


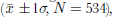 the oxalate has a background level <2 ng g
the oxalate has a background level <2 ng gIntroduction
Oxalate ((COO)2 2–) is the chief atmospheric dicarboxylic compound (Reference Kawamura and IkushimaKawamura and Ikushima, 1993; Reference Kawamura and IkushimaKawamura and Usukura, 1993), and is ubiquitous in the troposphere (Reference Schuetzle, Cronn and CrittendenSchuetzle and others, 1975; Reference Cronn, Charlson, Knight, Crittenden and AppelCronn and others, 1977; Reference Grosjean, van Cauwenberghe, Scvhmid, Kelley and PittsGrosjean and others, 1978; Reference Norton, Roberts and HuebertNorton and others, 1983; Reference Yokouchi and AmbeYokouchi and Ambe, 1986; Reference Kawamura and Gagosian.Kawamura and Gagosian, 1987; Reference Kawamura and UsukuraKawamura and Kaplan, 1987; Reference Satsumabayashi, Kurita, Yokouchi and UedaSatsumabayashi and others, 1989, Reference Satsumabayashi, Kurita, Yokouchi and Ueda1990; Reference Kawamura and UsukuraKawamura and Usukura, 1993). Oxalate may act as a kind of cloud-condensation nucleus, play an important role in geochemical processes in the atmosphere during a long-range transport of inorganic aerosols, and be potentially important to sea-water chemistry by controlling the vertical distribution of oxalic and other organic acids in the surface water (Reference Kawamura and UsukuraKawamura and Usukura, 1993). Oxalate presents in the atmosphere mainly in the form of particulates (Reference Norton, Roberts and HuebertNorton and others, 1983; Reference Kawamura and KaplanKawamura and Kaplan, 1987; Reference AndreaeAndreae and others, 1988; Reference Ludwig and KlemmLudwig and Klemm, 1988; Reference Sempere and KawamuraSempere and Kawamura, 1994; Reference Matsumoto, Nagao, Tanaka, Miyaji, Iida and IkebeMatsumoto and others, 1998; Reference Baboukas, Kanakidou and MihalopoulosBaboukas and others, 2000), while a fraction of it dissolves in cloud droplets (Reference Norton, Roberts and HuebertNorton and others, 1983; Reference Kawamura, Ng and Kaplan.Kawamura and others, 1985; Reference Sempere and KawamuraSempere and Kawamura, 1994), and an even smaller fraction appears in gaseous form (Reference Norton, Roberts and HuebertNorton and others, 1983; Reference Ludwig and KlemmLudwig and Klemm, 1988; Reference LeferLefer and others, 1994; Reference Baboukas, Kanakidou and MihalopoulosBaboukas and others, 2000). Although not certain at present, several sources have been found contributing to its atmospheric concentration. Provenance for direct emissions includes forest fires (Reference AndreaeAndreae and others, 1988; Reference LeferLefer and others, 1994; Reference Legrand and Angelis.Legrand and de Angelis 1995, Reference Legrand and Angelis.1996), motor exhaust and soil release (Reference Kawamura and KaplanKawamura and Kaplan, 1987; Reference GrosjeanGrosjean, 1989). Atmospheric reactions of anthropogenic hydrocarbons (Reference Grosjean, van Cauwenberghe, Scvhmid, Kelley and PittsGrosjean and others, 1978; Reference Grosjean and Friedlander.Grosjean and Friedlander, 1980; Reference Hatakeyama, Tanonaka, Weng, Bandow, Takagi and AkimotoHatakeyama and others, 1985, 1987) and biogenic unsaturated fatty acids (Yokouchi and Ambe, 1986; Reference Kawamura and Gagosian.Kawamura and Gagosian, 1987) are secondary sources. More recently, it has been suggested that volcanic eruptions also emit oxalate to the air (Reference Baboukas, Kanakidou and MihalopoulosBaboukas and others, 2000).
Past atmospheric variations of oxalate can be retrieved in ice cores from polar areas or mid- and low-latitude glaciers, given a good knowledge of the relation between the composition of snow and that of the atmosphere. Unfortunately, there have been only a few studies involving the record in polar ice cores (Reference Kawamura and YasuiKawamura and Yasui, 1991; Reference Legrand, de Angelis, Staffelbach, Neftel and StaufferLegrand and others, 1992; Reference Legrand and Angelis.Legrand and de Angelis, 1995, Reference Legrand and Angelis.1996), and even research on mid- and low-latitude regions is rare (Kang and others, 2001). Recent studies indicated (COO)2 2– in Greenland ice cores over the past 200 years averaged about 0.4 ng g–1, with perturbations probably reflecting biomass-burning events in the high northern latitudes (Reference Legrand and Angelis.Legrand and de Angelis, 1995, Reference Legrand and Angelis.1996). By contrast, in Far East Rongbuk Glacier (FER Glacier) (27˚59’N, 86˚55’ E), which is located about 13 km north of the peak of Qomolangma (Mount Everest), (COO)2 2– in the past 180 years averaged 13.7 ng g–1, with the highest values occurring in the 1960s. the concentration increase from the beginning of the 20th century has been attributed to industrial pollution (Kang and others, 2001).
In this paper, we investigate variations of the past 43 year oxalate record in an ice core from Ürümqi glacier No. 1 (UG1), a mid-latitude glacier in China, and compare the record with that of FER Glacier. We examine the possible implications for the oxalate source as well as the longitudinal atmospheric interaction between the Tien Shan and Qomolangma areas.
Ürümqi Glacier No. 1 and the Ice Core
Located at 43˚06’ N, 86˚49’ E in central Eurasia, UG1 is a valley glacier at Ürümqi river head, Xinjiang Autonomous Region, China. It is composed of east and west branches and occupies a total area of about 1.84 km2 (Liu and others, 1991) (Fig. 1). the equilibrium-line altitude averaged 3976ma.s.l. during 1979–89, and has risen to >4000ma.s.l. in recent years (Chinese Academy of Sciences, 1979–98). the mean annual precipitation at 4050ma.s.l. in the accumulation area of the east branch is 645.8 mma–1w.e. (Wang and Zhang, 1985; Yang and others, 1988). the May–September period accounts for 88% of the annual precipitation (Wang and Zhang, 1985; Yang and others, 1992; Reference Yinsheng, Kang and ChaohaiZhang and others, 1994). There are only a few cities or towns within 100 km of the glacier. the nearest town, Houxia, is located 50 km away in the river valley, and has a steel works and a cement plant which have been built since 1958. to the northeast of UG1,105 km away, lies Ürümqi, the capital city of Xinjiang.
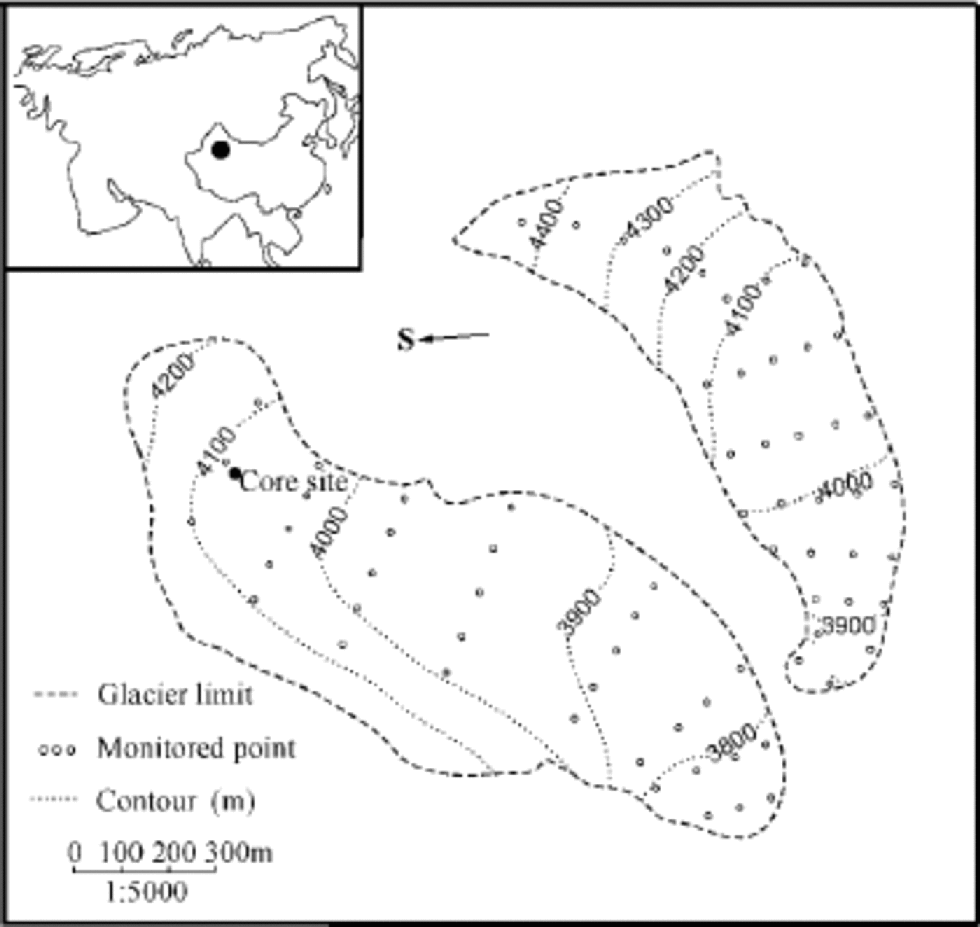
Fig. 1 UG1, with its location represented by the black dot in the inset map of Eurasia. the drilling site is located near the monitored point H2’ at the east branch of the glacier.
The ice core studied here is 14.08 m long and was retrieved in October 1998 at 4040 ma.s.l. in the superimposed-ice zone of the east branch of UG1, close to the monitored point H2’ at 4042ma.s.l. (Fig. 1). It is composed of clear, dense ice with scarce bubbles, which formed in the superimposed-ice zone, and opaque, bubbly ice with spherical bubbles 1–2mm in diameter, which formed in the percolation zone, as well as dust layers, of which two strong ones occurred at 0–6 and 1300–1317 cm, respectively. the superimposed ice accounts for 80–90% of the core, whereas the bubbly ice and the dust layers total 10–20%. In the upper part of the ice core, from 138 to 386cm, there is a nearly vertical crack. the ice within 2–3 cm of the fracture differs from the adjacent ice layers in its transparency and uniformly elongated bubbles, with long axes perpendicular to the fracture plane. It was formed from surface meltwater during the development of the crack (Lee and others, 2002a).
The core was cross-dated by δ18O, β activity, and variations of pyruvate and electric conductivity measurement (ECM). Temperatures from June to August control the δ18O in UG1 (Hou and others, 2000), so we dated the core first by comparing δ18O with the temperature in the area. This provides four reference dates: 1962,1966,1970 and 1976. Measurements of β activity provide events in 1963,1967,1976,1980 and 1986, corresponding to the known nuclear fallout events in the Northern Hemisphere and western China and to the Chernobyl nuclear accident. Dating via these two series differs by about 1 year. the whole core was dated by cross-counting peaks of ECM and pyruvate while consulting the reference dates from δ18O and β activity. It covers a period of 43 years from 1955 to 1998 with an uncertainty of about 1year (Fig. 2).
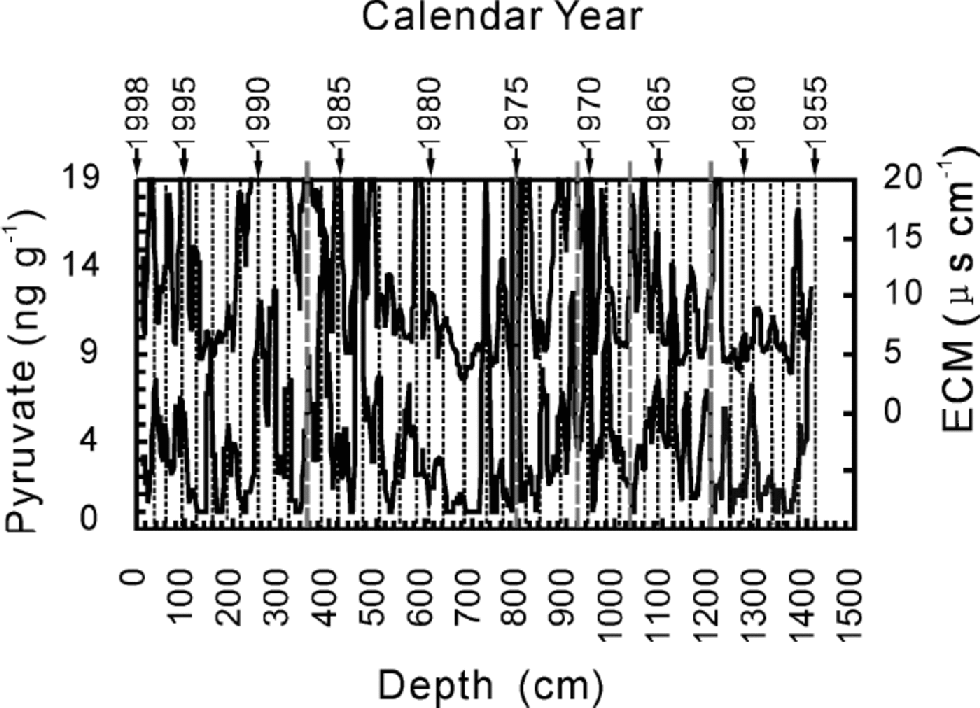
Fig. 2 Cross-dating for the whole ice core by ECM and pyruvate from the bottom (1955) to the surface. Solid straight lines protruding from the upper boundary of the graph indicate the reference levels from δ18O and β activity, and represent from right to left 1962, 1966, 1970, 1976 and 1986.
Analytical Techniques
The ice core was sectioned every 2 cm in the laboratory, and only the central part was used for analysis. In the cracked section, however, the analytical samples were selected to avoid the fracture as much as possible. In order to avoid the outer 1cm of the core that is susceptible to drilling contamination (Reference Legrand, de Angelis and MaupetitLegrand and others, 1993), the sampling was compromised by including parts of the transparent ice. the samples were kept frozen in glass vials with airtight covers until analysis.
Oxalate in 662 samples was analyzed by a DX-300 ion chromatograph with AS4A and ASRS-II columns. Gradient elution by Na2B4O7 and chemical suppression mode with 25 m MH2SO4 as well as a preconcentration technique were used for the measurement. A relative uncertainty of 4% for oxalate was obtained with a 2mL sample and calibrated by the ![]() Reference Standard IC-OXAL1X-1. for more details of the analysis, see Lee and others (2002b).
Reference Standard IC-OXAL1X-1. for more details of the analysis, see Lee and others (2002b).
Results and Comparison
Oxalate is a minor organic compound compared with acetate and formate, the light carboxylic acids which in the ice core average 392.3±390.8ng g–1 and 102.8±147.3 ng g–1
![]() N = 662), respectively (Lee and others, 2001). Although the background level is about 1–2 ng g–1, oxalate concentration in parts of the ice core is below the detection limit (see upper graph in Fig. 3). If the concentration in the crack, where (COO)2
2– is significantly enhanced (shaded area in the upper graph of Fig. 3) due to the cracking effect (Lee and others, 2002a), is not taken into account, the mean concentration of oxalate in the past 43 years is 3.6±9.2 ng g–1 (N = 534), nearly 10 times higher than in the Greenland ice cores (Reference Legrand and Angelis.Legrand and de Angelis, 1995). In the FER Glacier ice core, however, (COO)2
2– averages 23.7±41.1ngg–1 (N =216) from 1955 to 1996 (Kang and others, 2001), about 6 times higher than the mean concentration in UG1.
N = 662), respectively (Lee and others, 2001). Although the background level is about 1–2 ng g–1, oxalate concentration in parts of the ice core is below the detection limit (see upper graph in Fig. 3). If the concentration in the crack, where (COO)2
2– is significantly enhanced (shaded area in the upper graph of Fig. 3) due to the cracking effect (Lee and others, 2002a), is not taken into account, the mean concentration of oxalate in the past 43 years is 3.6±9.2 ng g–1 (N = 534), nearly 10 times higher than in the Greenland ice cores (Reference Legrand and Angelis.Legrand and de Angelis, 1995). In the FER Glacier ice core, however, (COO)2
2– averages 23.7±41.1ngg–1 (N =216) from 1955 to 1996 (Kang and others, 2001), about 6 times higher than the mean concentration in UG1.

Fig. 3 Comparison of oxalate variation in the UG1 ice core (uppergraph) with the record from FER Glacier. Both curves result from the six-point running mean of their data. the strong shaded peak in UG1 results from post-depositional effect of the crack.
Despite its low average concentration, oxalate does have spikes, most of which reach 10–30 times higher than the background level and cover a period of approximately 1 year. Striking peaks from right to left (Fig. 3) correspond to 1962, 1966, 1970, 1973, 1978, 1979, 1983 and 1986. These spikes are not necessarily related to the percolation ice layers nor to the dust ones. the two strong dust layers, for instance, are both free of the oxalate peaks. Therefore the oxalate increases are independent of the constitution of the ice, as opposed to the effect of the cracking.
Kang (1999) dated the FER ice core using β activity, δ18O and profiles of Ca2+ and SO4 2–.The reference dates 1954 and 1963 are at 9.4 and 6.9m, respectively. the years from 1955 to the core surface in 1996 stretch a length of 9m (Kang, 1999). Based on the dating, the spikes in UG1 match those in FER Glacier during the past 40 years: most of the spikes in one glacier have counterparts in the other, especially allowing for dating errors for the two cores (Fig.3). Note, however, that the strong shaded peak in UG1 is caused by the crack, and thus has no corresponding peak in FER Glacier. on the other hand, peaks in FER Glacier are much higher than, although not strictly proportional to, their counterparts in UG1, which probably indicates a greater increase of oxalate in the atmosphere over FER Glacier. Peaks in FER Glacier are also wider than those in UG1. the spatial interval for analytical samples in the FER core is 4 cm. Given the glacier’s mean annual accumulation of 22 cm a–1 ice equivalent in the past 40 years, the average sample resolution is 5.5 samples per year compared to 16.8 samples per year for UG1. the lower resolution of sample selection in the FER core would certainly widen the peaks. the wider peaks in FER Glacier may also be related to the possibility that the concentration enhancement in the atmosphere dominates for a longer time over the Qomolangma area, whereas it both increases and decreases quickly and lasts a shorter time in the Tien Shan. the relative importance of the two possibilities cannot be assessed here because insufficient data are available.
The oxalate records in the two areas can be divided into four periods in terms of their concentration during the past 40 years (Table 1). UG1 recorded low levels of oxalate in the latter half of the 1950s, with a mean concentration of 2.5 ng g–1, whereas the 1960s and 1970s saw the highest average concentration of 5.0 ng g–1, with the peaks occurring mainly in the latter period. Oxalate levels in the 1980s are close to those in the later 1950s, with an average concentration of 3.0 ng g–1 resulting chiefly from the spike in 1986. the 1990s have the lowest concentration, which is below the detection limit for most samples. As in the UG1 core, oxalate in the FER ice core also has the highest concentration in 1960–80, and the lowest concentration in the 1990s, with a middling concentration in 1955–60 and the 1980s (see Table 1). the average concentration of 24 ng g–1 in the 1980s is also mainly due to the 1986 peak.
Table 1. Mean oxalate concentration (in ng g–1) recorded in UG1 and FER Glacier during periods 1955–60, 1960–80, 1980–90 and 1990–96

Discussion
The correlation of oxalate records between UG1 and FER Glacier suggests that (COO)2 2– over the two areas may have varied synchronously or had a common source.
The co-variation of (COO)2 2– may be caused by local sources which have been in phase with each other in releasing oxalate to the atmosphere. the oxalate flux, however, differs, being much larger at FER Glacier than at UG1. It is probably this difference that causes oxalate peaks to be stronger in FER Glacier than in UG1. the timing of the peaks, on the other hand, is the same, which suggests that the two areas may have had the same kind of local sources.
The other plausible explanation is that the two areas have shared the same source, and the Indian subcontinent may be the main provenance for both records. This scenario is based on the longitudinal atmospheric circulation linking the two areas. Although some meteorologists doubt the linkage (e.g. personal communication from Qian Zhengan, 2000), airmass exchange by the circulation has been observed. Schematically shown in Figure 4 is an example of the circulation along 90˚E on 1 June 1979 (Reference Zhang, Ruihua, Dali and ShuhaiZhang and others, 1984). to the south of the Qinghai–Tibetan Plateau, Hadley circulation dominates. the upwelling air mass develops from south of 15˚N and turns north in the upper level of the troposphere. It crosses over the plateau and then descends around 50˚N near the Tien Shan. This longitudinal circulation is prone to occur in winter and spring when the Indian monsoonal circulation system is not active, and when oxalate has higher concentration in precipitation over UG1, as discussed below. the longitudinal circulation acts as a conveyer for chemical species from the tropical area to reach the mid-latitudes. In a recent study, it was found that oxalate can be transported over a long distance due to its existence in fine particles (Reference Matsumoto, Nagao, Tanaka, Miyaji, Iida and IkebeMatsumoto and others, 1998). This indicates the feasibility of (COO)2 2– being transported from the Indian subcontinent to the Tien Shan. the gradual oxalate depletion of the air mass due to deposition in the long transportation could result in the lower oxalate in UG1.

Fig. 4 An example of longitudinal air movement along 90˚ E on 1 June 1979, demonstrating the direct atmospheric connection between the Tien Shan and Qomolangma areas. Ha stands for Hadley circulation. Modified from Reference Zhang, Ruihua, Dali and ShuhaiZhang and others (1984).
The geographical contrasts make a chemical connection between the two sectors worthy of attention. UG1 is located in the central part of the Eurasian continent. It is surrounded by vast areas of deserts and the Gobi, whereas FER Glacier lies below 30˚N. the glaciers are separated longitudinally by 41600 km in very different geographic environments. They also differ considerably in height. FER Glacier protrudes two-thirds of the way through the troposphere, and the drilling site is 2460 m higher than that at UG1. Additionally, the sites differ in the regional prevailing atmospheric circulation systems (Reference Dai and JiaxiDai,1990). However, the oxalate-record correlation suggests that atmospheric connection between the two areas is stronger than previously thought.
Unfortunately, our limited dataset does not allow a more definitive explanation for the correlation. the primary source of oxalate, however, can be pinpointed as anthropogenic as follows. In contrast to the Greenland ice-core record, where spikes of oxalate result from boreal forest fires (Reference Legrand and Angelis.Legrand and de Angelis, 1995, Reference Legrand and Angelis.1996), the concentration enhancements in UG1 are unlikely to be from forest fires, since no forest fires intense enough to affect the surrounding atmosphere have been observed in either western China or the Indian subcontinent corresponding to the years when the spikes occurred. Biogenic emission is not the main source either. Because the coverage of vegetation in western China is very limited, emission from vegetation would not cause significant changes in atmospheric concentration. As mentioned above, precipitation in seasons other than summer over the UG1 area accounts for only about 22% of the whole year, which means a very low frequency of precipitation in these seasons (especially winter). A recent study showed that oxalate tends to concentrate in precipitation events of lower frequency, partly because a high precipitation rate keeps oxalate low in the atmosphere by scavenging it frequently, causing a low oxalate concentration for individual precipitation events (Reference Sempere and KawamuraSempere and Kawamura, 1994). This mechanism explains the oxalate enhancements in UG1 during winter. Since this is not the growth season, the oxalate spikes cannot be mainly the result of vegetational emission from the Indian subcontinent, nor are they likely to be due to emission of soil. the primary provenance appears to be directly or indirectly from anthropogenic emission. This is partly supported by evidence from the FER ice core that the mean concentration of oxalate is 2–3 times higher in the 20th century than in the 19th century (Kang and others, 2001). for the synchronism, pollution from Ürümqi and Houxia and from the densely populated Indian subcontinent would act, respectively, as sources for each ice-core record. on the other hand, if the two areas have shared the same source, they both result primarily from anthropogenic emissions in the Indian subcontinent. In either case, the oxalate variations coincide with recent industrial/economic development in southern Asia. the period of relatively low concentration before 1960 through to the highest concentration in the 1960s and 1970s corresponds to the process of regional economic growth, while the decline in the 1980s and then eventually to the background in the 1990s may reflect the measures prompted by governmental and industrial awareness of the environmental problem during the past 20 years.
Conclusions
The past 43 year oxalate record in an ice core from UG1 shows that oxalate is a minor carboxylic species with a mean concentration of 3.6±9.2 ng g–1
![]() The background level is 52 ng g–1, with some samples having a concentration below the detection limit. the record is nearly 10 times higher than in Greenland ice cores, but about 6 times lower than in FER Glacier.
The background level is 52 ng g–1, with some samples having a concentration below the detection limit. the record is nearly 10 times higher than in Greenland ice cores, but about 6 times lower than in FER Glacier.
Superimposed on the background level of the UG1 record are sporadic oxalate enhancements, which correlate with those in FER Glacier in the past 40 years. This suggests that the two areas may have had the same kind of local sources, with the source strength for FER Glacier much larger than for UG1, or, alternatively, a common source from the Indian subcontinent via the longitudinal circulation of atmosphere. In either case, the oxalate record coincides with industrial/ economic development in southern Asia, and is directly or indirectly due to anthropogenic emissions.
Acknowledgements
We would like to thank Hou Shugui, Xiao Cunde, Tian Lide and He Yuanqing of CAREERI for helpful discussions, and Qian Zhengan and Li Peiji of the same Institute for their reviews of an initial draft of this paper. We acknowledge valuable suggestions and editorial assistance from UT. van Ommen, and comments from two anonymous reviewers that helped improve the clarity and accuracy of the paper. This study was financed by the National Natural Science Foundation of China (grant Nos. 40073035 and 49871022), the Chinese Academy of Sciences (grant No. KZ951-A1-402-03), the Tien Shan Glaciological Station and the Chinese postdoctoral research foundation.





