


Introduction
Over the last few years, there has been growing interest in the study of the polar ice sheet, mainly with the aim of reconstructing the interrelationships between the chemistry of the atmosphere and the paleoclimate. Moreover, studies on the mass balance of the Antarctic ice sheet are very important for our understanding of the interrelations between global warming and sea-level changes.
The chemical composition of snow and firn samples from Campbell Glacier, northern Victoria Land, Antarctica, was studied to investigate seasonal signal and chemical contribution from some emission sources (marine biogenic activity, sea and crust). New data on the snow-accumulation rate of Campbell Glacier will improve our evaluation of the mass balance of this glacier.
Sampling and Analysis
A snow pit (2 m deep) was dug in the lower part of the Campbell Glacier tongue (site A: 74°41 S, 164°30' E), about 6.5 km from the cliff, at 50 m a.s.l., during the 1994-95 Italian Antarctic Expedition. Another snow pit (1.4 m) was dug in Campbell Glacier at about 800 m a.s.l. (site B: 74°15'S, 164°04'E), just opposite Mount Campbell and about 50 km from the frontal ice cliff. A firn core (2.37 m) was collected at the bottom of this pit. Finally, another core (7 m) was obtained at 1560 m altitude from the upper sector of this glacier, not far from the confluence of the tributary Recoil Glacier, and about 110 km from the ice cliff (site C: 73°45' S, 163°20' E). Figure 1 shows the sampling map.
Site A, at the glacier tongue, is situated in the percolation zone, where surface melting occurs in the summer. The pit dug in this area revealed many ice layers and pipelike structures as ice glands. Sites B and C are located in the dry-snow zone of Campbell Glacier, as demonstrated by the temperatures at 3.5 m depth, well below -20°C Melting does not usually occur here.

Fig. 1. Map of Campbell Glacier showing sampling sites.
Samples were stored frozen. Then, following surface cleaning in a cold room, they were subsampled and stored in pre-cleancd (with 18 Mohm ultra-pure water) polyethylene containers and sent to the laboratory where they were melted in a clean room prior to the chemical and isotopic analyses.
Density was calculated weighing a measured volume of the snow or firn subsamples analyzed.
Analyses of Na+, K+, Mg2+, Ca2+, CH3SO3- −, Cl−, NO3 − and SO4 2- were performed by ion chromatography (Dionex 2020i). Anion separation was obtained by means of a Dionex AS5 column, according to the procedure outlined by Legrand and others (1993). Cation separation was performed using a Dionex CS12 (methane sulfonic acid (MSA) 20 mM eluent). The eluent flow was 1 ml mill−1 and the sample volume was 1.5 ml.
Table 1. Concentrations of studied samples
H2O2 was analyzed by an electrochemical detector (ANTEC mod. “Decade"), equipped with a micro flowcell (volume 0.005 μl) with a Pt working elecirode (diameter 0.5 mm) in which the potential of the cell was 650 mV. The detector was on line with a pump and Chromatograph injection valve. The flow rale of the mobile phase (0.01 M Na2HPO4-0.01 N KH2PO4 solution) was 0.4 ml min −1 and the loop was 100 μl.
The relative repeatability, for all chemical species analyzed, was better than 10%.
The determination of the oxygen isotope composition was carried out according to the technique of isotopic equilibration of CO2 with water, using an automatic equilibration device (Isoprep 18) on line with an Optima VG mass spectrometer.
The results are reported as δ units per mil (‰) where

R being the l8O/l6O. The δ18O values are reported vs Vienna Standard Mean Ocean Water isotopic standard. The standard deviation of oxygen measurements for the automatic preparation device was equal to an average of ± 0.10‰.
Results and Discussion
Chemistry of firn the mean concentration of the ions analyzed is given in Table 1. The concentration of the elements derived from sea salt (typically Na+, Mg2+ and Cl−) decreased rapidly from the coast to the higher altitudes. This feature has already been described for different Antarctic sites by Reference Herron and LangwayHerron and Langway (1979), Legrand and Reference Delmas, Legrand, Aristarain and ZanoliniDelmas (1985), Mulvaney and others (1993),Reference Minikin, Wagenbach, Graf and KipfstuhlMinikin and others (1994) and, in the case of northern Victoria Land, by Piccardi and others (1994, 1996) and Reference CaprioliCaprioli and others (1997).
The higher sea-salt concentration resulting for the Campbell Glacier tongue, about 6 km from the ice edge, is mainly due to dry deposition of large particles from sea-salt aerosol during sea storms (Fig. 2). This process has been observed, for example, at the Filcher Ronne Ice Shelf (Minikin andothers, 1994). The event recorded in the firn layer at about 40 cm depth at site A could be attributed to the storm that struck the Ross Ice Shelf and then moved toward the Transantarctic Mountains at the beginning of June 1993. The wind storm was so severe that some of the automatic weather stations on the Ross Ice Shelf were damaged (Stearns and others, 1994).
In the analysis of the correlation between Na+ and Cl− . the equations of the regression lines were Cl− μeql−1 = 2.02 + 0.99Na+ μeql−1 (r = 0.99, n = 156) for site C, and Cl− μeq 1−1 = 6 + 1.07Na+μeql −1 (r = 0.98, n = 85) for she B, both near the dilution line of bulk sea water (CT−/Na+ = 1.17). These results suggest, over the overall dataset, an absence of significant fractionation of the reaction products (HCl and Na2SO4) between NaCl and H2SO4 (Legrand and Delmas, 1988). Nevertheless, some samplesshowed a significantly higher or lower CL/Na+ ratio with respect to the Cl−/Na + ratio in sea water.This confirms that in particular conditions fractionation of HCland Na2SO4 may occur.
The equation of the regression line resulting for site A was Cl− µeql−1 = 239 + 0.72Na+µeq1−1(r = 0.95, n = 21), indicating a significantly lower Cl /Na+ ratio with respect to the sea water. This is due to an enrichiment of Na2SO4 as shown from data obtained from snow samples collected at about 35 cm depth. The nssSO4 2- profile nssSO4 2- = SO4 2-” -0.12Na+ , where all species are in µeql −1) at site A is provided in Figure 2.
The mean Na+/Mg2+ ratios were 5.1,4.4 and 5.4 for sites A, B and C, respectively, which are close to the sea-water Na+/Mg2+ ratio (4.14) according to Reference Ikegami, Zaizen and MakinoIkegami and others (1994) and Reference Mclnnes, Covert, Quinn and GermaniMclnnes and others (1994). They found no fractionation of Mg2+relative to Na+ in aerosol collected from a remote marine boundary layer or the upper tropical troposphere. Nevertheless, in some samples an Na+/Mg2+ ratio higher than the sea-water ratio was observed, suggesting sporadic depletion of Mg2+ relative to Na +.This is in keeping with the results ofMulvaney and others (1993), in a study of the fractionation of sea salt during transport across an Antarctic ice shelf.
Generally speaking, the Na+/K+ and Na+/Ca2+ ratios weremuch lower than the bulk sea-water ratios at sites B and C.The percentages of nssK+ (nssK+ = K+- 0.0213Na+, where all species are in µeql−1) and nssCa (nssCa2+ = Ca2+-0.043 Na+, where all species are in μeql−1) with respect to the total content of these ions were 72.8% and 81.8% at site B and 82.6% and 81.4% at siteC. These percentages, which are comparable to those reported by Whitlow and others (1992) at the South Pole, confirm a prevailing crustail origin of K+ and Ca2+.
The MSA, produced in photo-oxidation of dimethyl sulfide, which is, in turn, derived from the biologically produced precursor, dimethyl sulfonipropionate (Reference Dacey and N. V.Dacey and Blough, 1987), showed mean concentrations (0.21 μM) consistent with data obtained at other Antarctic sites (Key and others, 1986; Saigne and Legrand, 1987; Prospero and others, 1991; Legrand and others, 1992; Mulvaney and others, 1992; Minikin and others, 1994). If the data for the Campbell Clacier tongue are OOt considered, because of ihr low number of samples analyzed (12), in which winter samples with lower MSA content could prevail, a decreasing concentration of MSA is observed as altitude increases. The mean concentrations of MSA were 0.27 and 0.17 μM for siles B and C, respectively.
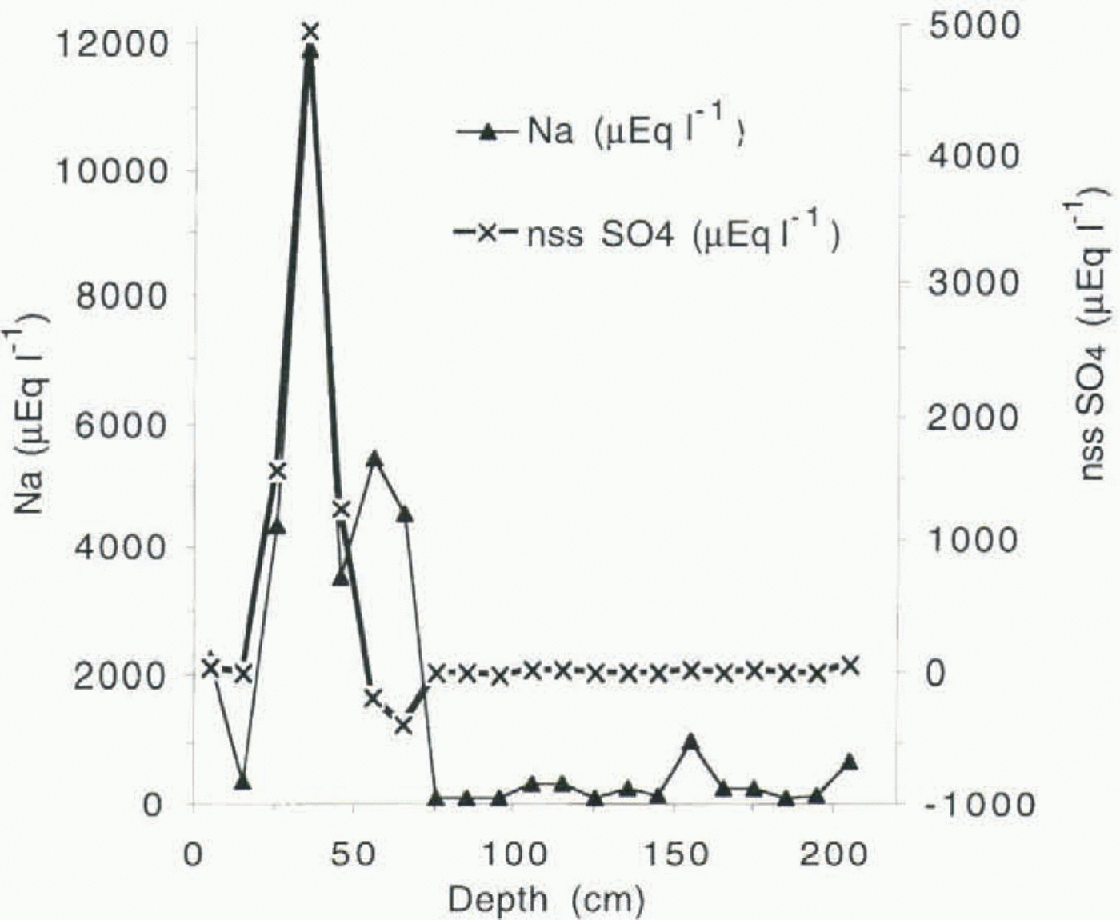
Fig. 2. Na+ and nssSO4 2- profile at site A.
NO3 is produced in the oxidation of NOx. in Antarctica, ihe most important sources of NOx are lightning ano1 transport from the stratosphere troposphere. Soil release, biomass burning, fossil-fuel burning and volcanic emission are not considered significant sources of NOx (Herron; 1982; Legrand and Delmas, 1986).
The mean concentrations of NO3 ranged between 2.1 µeql−1 (site A and 0.97 µeq1−1 (site C). These values are consistent with the data reported for other Antarctic sites (Neubauer and Heitmann, 1988; Legrand and Kichner, 1990; Qin Reference Dahe, Zeller and DreschhoffDahe and others, 1992; Whitlow and others, 1992; Minikin and others, 1994; Piccardi and others, 1994; Caprioli and others, 1997).
The contribution of sulfate in the Antarctic region is mainly due to marine biogenic activity and sea salt. Crustal and antropic sources are generally negligible. Volcanic events can produce a significant input of SOx into the atmosphere, which can increase SO4 2 deposition and yield a very marked signal in the snow, at sites either close to or far from eruptions (Reference HammerHammer, 1977, Reference Hammer1985; Delmas and others, 1985; Reference Moore, Narita and MaenoMoore and others, 1991).
In our samples, the mean nssSO4 2- /SO4 2- ratios were -0.055,0.54 and 0.42 at sites A, B and C, respectively. Negative values of nssSO4 2 can be found (especially at coastal polar sites and in winter) due to little-known processes that lead to an SO4/Na+ ratio lower than that of bulk sea water. The low nssSO4 2-/SO4 2- ratio at site A confirms the strong marine influence that decreases sharply away from the sea. The nssSO4 2- correlation washighly significant(r =0.88) n = 85) for site B, but less significant for site C (r = 0.26, n = 156). Moreover, the relationship between Ca2+ nssCa2+, SO4 2- and nssSO4 2- at sites B and C differed considerably. At site B, nssSO4 2- and nssCa2+ are significantly correlated (nssCa2+ = 2.36 + 0.92nssSO4 2; r - 0.84), and a large part of the nssCa2 + could be in CaSO4 form, whereas at site C, nssSO4 2 and nssCa2+ were not correlated (nssCa2+ = 3 + 0.33nssSO4 2; r = 0.08) (Fig. 3). The nssCa2+/nssSO4 2- ratios (excluding the negative values of nss SO4 2- were 1.41 and 3.73 at sites B and C, respectively, due to a relatively lower content of nssSO4 2- with respect to other constituents at site C. This low content of nssSO+ 2- is also perceptible in other ratios such as MSA/nssSO4 2- (0.092 and 0.26 at sites B and C,respectively).
H2O2 is produced in the atmosphere by photochemical processes which reach their maximum intensity during the summer months. Therefore, the atmosphere and snow show a peak in H2O2 concentration during this period. Due to this pattern, H2O2 is used as a seasonal tracer. The mean concentration of H2O2 was 8.45and 8.5 µg1−1 at sites B and C, respectively. This suggests that the deposition processes at these two sites do not differ.
In order to explain the relationship between the elements and compounds studied from sites C and B,chemical fractionation and/or reaction inside air masses, and the different origin of the air masses at the two sites should be considered. The peculiar chemical characteristics of the firn at sitesB and C, and particularly the sharp decrease in nssSO4 2- cannot be justified by the fractionation of chemical species alone. Thus, the contribution of masses of air of different origin should be taken into consideration for sites B and C.
Stable-isotope analyses the paleotemperalure profiles derived from the δ18O and δ data obtained from thelong ice cores obtained in Greenland and Antarctica, are long-standing evidence of the importance of determining the stable-isotope composition of ice cores. in fact, as initially observed by Reference DansgaardDansgaard (1964), oxygen and hydrogen isotopic values are related to several meteorological parameters such as the condensation temperature, the cooling processes (i.e. isobaric or adiabatic), the initialisotopic composition of vapor and the trajectory followed by the air masses. As a consequence, linear relationships have been observed in different Antarctic areas linking the mean annual isotopic composition of precipitation to the mean annual surface temperatures (Reference Lorius and MerlivatLorius and Merlivat, 1977; Peel and Clausen, 1982; Reference Isaksson and KarlénIsaksson and Karlen, 1994). Decreasing values of the isotopic composition would be observed as one moves inland throughout Antarctica, according to the above considerations.
The stable-isotope composition profile of a firn core can be seen as a temporal record of data thatreach their least negative values in summer and their most negative ones in winter, allowing one toestablish a chronological scale and to evaluate the mean accumulation rate characterizing the area during the time period considered. in areas where the accumulation rate is very low, homogenizationprocesses may play an important role in the uppermost part of the snow column, as pointed out by johnsen (1977), totally or partially obliterating the pristine seasonal signal. This is particularly true inland on the Antarctic ice sheet. Obviously, melting at the surface and percolation through the snow-pack and refreezing may also be important factors. Another external factor which can contribute to the obliteration of the original signal is the redistribution of the snow by the wind, which can be very effective in some Antarctic regions. Nevertheless, a tentative chronological scale can be established whenever all these phenomena seem to be less evident.
At site A, δ18O values ranged between -18.2‰ and -32.9‰, with a mean value of-23.9‰. Thealmost complete obliteration of the seasonal signal at this low-elevation site may be due to surface melting occurring in summer. As regards the intermediate-elevation site B, the isotopic values ranged from -23.5‰ to -35.9‰, with a mean value of -27.2‰. in addition, the seasonal signal did not appear to be well preserved in this case and it is quite difficult to expla in the smoothed profile by melting processes occurring at the surface in view of the preservation of the chemical seasonal signal in the same core. The explanation may be a redistribution created by the wind, or perhaps a problem with the storage of the core samples. Only one of the chemical parameters, H2O2 exhibited a similar pattern, which may support the second hypothesis.

Fig. 3. nssSO4 2--nssCa2+ correlations at sites B and C
A totally different situation was observed in the case of the higher-elevation site C. Here the seasonal signal record was fairly well preserved, the least negative values being ascribed to the summer precipitation and the most negative ones to the winter period. The δ18O values rangedfrom -27.2‰ to -40.8‰, with a mean value of -33.5‰. The variation of the oxygen-isotope compositionwith depth is shown in Figure 4, along with the chemical profiles.
Comparing the mean δ18O values for the three cores obtained at different altitudes, and assuming these values represent approximately the mean δ18O of precipitation at each site, a mean isotopic vertical gradient of -0.6%(100m)−1 can be observed along the glacier axis (0.4‰ (100 m) −1 between 50 and 800 m, and -0.8‰ (100m)−1 between 800 and 1560 m). Similarly, a mean “continental” gradient of-0.1‰km−1 was obtained, the more negative values being found inland. These gradients are acceptable and lie with in the range of values already reported in the literature (Dansgaard and others, 1973; Qin Dahe and others, 1994); they were previously measured by the authors in the Terra Nova Bay area. Using the Lorius and Merlivat (1977) relationship, δ18O = 0.755t(°C)-7.6 (valid for East Antarctica), a mean temperature-elevation gradient of -0.8°C (100 m)−1 may be calculated.

Fig. 4. Profiles of chemical species at site B.

Fig. 5. Profiles of isotopic and chemical species at site C.
Seasonal Trend of Chemical Species
Unfortunately, the isotopic and H2O2 profiles obtained at sites A and B cannot be used lor dating purposes due to partial obliteration of the seasonal signal.
Figure 4 shows the chemical profiles for MSA, NO3 − and nssSO4 2 at site B, and Figure 5 shows the profiles for δ18O, H2O2 MSA, nssSO4 2- and NO3, at site C. All species showed numerous well-defined annual cycles in which the δ 18O, H2O2, MSA, nssSO4 2 and NO3 peaks correspond to the late-spring-summer period.
When using chemical species profiles to date snow layers, some of the greatest difficulties are related to the in-accurate overlapping of the different profiles. T0 solve this problem, Udisti (1996)suggests a method involving a sum of normalizing profiles. in this study, we have restricted the data analysis, plotting depth vs chemical species concentrations by a cubic-spline function (figs 4 and 5) and then plotting depth, corresponding to different years for different chemical species, vs year (Fig. 6, for site C only :. in this manner, the differences emerging from the different chemical species profiles can be easily noted.
The number of summer peaks identified according to ihr chemical and isotopic profiles is given in Table 2.
Taking the mean density (site B = 0.46, site C = 0.384) into account, the mean accumulation rate was with in the range 150-170 kg m−2 a−1 at 800 m a.s.l., where 10 or 11 years of snow accumulation were identified, and with in the range 140-180 kg m−2 a−1 at 1560 m a.s.l., where 14-18 years of snow accumulation were identified. These values are in keeping with those found by Udisti (1996) in the Terra Nova Bay area, whereas Zanon (1989), on the basis of glaciological considerations, proposed an accumulation rate of 200 kg m −2 a−1 at the Camphell Glacier tongue.

Fig. 6. Chronological profile derived from the concentrations of different chemical species at site C.
Conclusions
The chemical composition of the snow and firn samples (rom Campbell Glacier confirms the decrease of sea-salt concentration with higher altitudes and increasing distances from the sea. The mean concentration οf Na+, for instance, fell sharply from 1750 μeq1−1 at site A (witha maximum of 11900 μeq1−1, an unusually high level, probably-due to a strong sea storm) to 39 and 21 μeq 1−1 at sites B and C, respectively, where the maximum values only slightly exceeded 210 μeq1−1.
Table 2. Number of summer peaks identified at sites B and C

The concentration of many of the studied elements and compounds (particularly δ18O, H2O2, MSA, nssSO4 and NO- , at site C) offers a good seasonal signal, showing higher content during the late-spring-summer period. It was therefore possible to identify many annual cycles (14-18 years for the 7 m firn core). By means of the measured densityvalues, the accumulation rate was also calculated for lower and upper Campbell Glacier. It did not differ much between the two sites (140-170 kg m 2 a−1 at site B and 140-180 kg m2 a−1 at site C) and is consistent with the rates previously calculated for the same area.
Acknowledgements
This work has carried out with the financial support of the Italian Programma Nazionale di Ricerchein Antartide.








